
Abstract
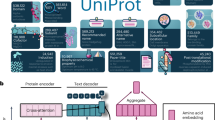
ProTrek unifies protein sequence, structure and natural language function in a trimodal language model through contrastive learning, enabling comprehensive searches between any two modalities, including within modality. ProTrek surpasses current alignment tools (for example, Foldseek and MMseqs2) in speed and accuracy for identifying functionally related proteins. Computational and wet-lab experimental validations show that the ProTrek server (www.search-protrek.com), with precomputed embeddings for over 5 billion proteins, efficiently processes and analyzes large-scale protein repositories.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


Data availability
The pretrained model weights (650 million and 35 million parameters) of ProTrek can be obtained online (https://huggingface.co/westlake-repl/ProTrek_650M and https://huggingface.co/westlake-repl/ProTrek_35M, respectively). All precomputed protein sequence, structure and text embeddings for the SWISS-PROT database are available online (https://huggingface.co/datasets/westlake-repl/faiss_index). Larger protein databases (TB-scale embeddings) are accessible through ProTrek’s web server (http://search-protrek.com) and official GitHub (https://github.com/westlake-repl/ProTrek), which contains billions of entries. The structural 3Di sequences are available from GitHub (https://github.com/steineggerlab/foldseek). The text descriptions are available from UniProt (https://www.uniprot.org).
Code availability
ProTrek is open-sourced under the MIT license. The code repository is available from GitHub (https://github.com/westlake-repl/ProTrek). The ProTrek web server can be accessed online (http://search-protrek.com). ColabProTrek v1 and v2 are available online (https://colab.research.google.com/github/westlake-repl/SaprotHub/blob/main/colab/ColabProTrek.ipynb and https://colab.research.google.com/github/westlake-repl/SaprotHub/blob/main/colab/ColabSeprot.ipynb?hl=en, respectively). For users requiring private database integration, we provide command-line tools to build embeddings for local deployment through GitHub (https://github.com/westlake-repl/ProTrek#add-custom-database).
References
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Zhang, Y. & Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 (2005).
Van Kempen, M. et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 42, 243–246 (2024).
Suzek, B. E. et al. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015).
Bileschi, M. L. et al. Using deep learning to annotate the protein universe. Nat. Biotechnol. 40, 932–937 (2022).
Gane, A. et al. ProtNLM: model-based natural language protein annotation. Preprint at https://storage.googleapis.com/brain-genomics-public/research/proteins/protnlm/uniprot_2022_04/protnlm_preprint_draft.pdf (2022).
Gligorijević, V. et al. Structure-based protein function prediction using graph convolutional networks. Nat. Commun. 12, 3168 (2021).
Zhou, N. et al. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biol. 20, 1–23 (2019).
Radivojac, P. et al. A large-scale evaluation of computational protein function prediction. Nat. Methods 10, 221–227 (2013).
Liu, W. et al. PLMSearch: protein language model powers accurate and fast sequence search for remote homology. Nat. Commun. 15, 2775 (2024).
Hong, L. et al. Fast, sensitive detection of protein homologs using deep dense retrieval. Nat. Biotechnol. 43, 983–995 (2025).
Achiam, J. et al. GPT-4 technical report. Preprint at https://arxiv.org/abs/2303.08774 (2023).
Touvron, H. et al. LLaMA: open and efficient foundation language models. Preprint at https://arxiv.org/abs/2302.13971 (2023).
Touvron, H. et al. LLaMA 2: open foundation and fine-tuned chat models. Preprint at https://arxiv.org/abs/2307.09288 (2023).
Guo, D. et al. DeepSeek-R1: incentivizing reasoning capability in llms via reinforcement learning. Preprint at https://arxiv.org/abs/2501.12948 (2025).
Ferruz, N., Schmidt, S. & Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 13, 4348 (2022).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Elnaggar, A. et al. ProtTrans: toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 44, 7112–7127 (2021).
Zhou, X. et al. Decoding the molecular language of proteins with Evolla. Preprint at bioRxiv https://doi.org/10.1101/2025.01.05.630192 (2025).
Peng, F. Z. et al. PTM-Mamba: a PTM-aware protein language model with bidirectional gated Mamba blocks. Nat. Methods 22, 945–949 (2025).
Su, J. et al. SaProt: protein language modeling with structure-aware vocabulary. In Proc. 12th International Conference on Learning Representations (ICLR, 2024); https://openreview.net/forum?id=6MRm3G4NiU
Su, J. et al. SaprotHub: making protein modeling accessible to all biologists. Preprint at bioRxiv https://doi.org/10.1101/2024.05.24.595648 (2024).
Radford, A. et al. Learning transferable visual models from natural language supervision. In Proc. 38th International Conference on Machine Learning (eds Meila, M. & Zhang, T.) 8748–8763 (PMLR, 2021).
Gu, Y. et al. Domain-specific language model pretraining for biomedical natural language processing. ACM Trans. Comput. Healthc. 3, 1–23 (2021).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370 (2003).
UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212 (2015).
Koehler Leman, J. et al. Sequence–structure–function relationships in the microbial protein universe. Nat. Commun. 14, 2351 (2023).
Todd, A. E., Orengo, C. A. & Thornton, J. M. Evolution of protein function, from a structural perspective. Curr. Opin. Chem. Biol. 3, 548–556 (1999).
Douze, M. et al. The Faiss library. Preprint at https://arxiv.org/abs/2401.08281 (2024).
Liu, S. et al. A text-guided protein design framework. Nat. Mach. Intell. 7, 580–591 (2025).
Xu, M., Yuan, X., Miret, S. & Tang, J. ProtST: multi-modality learning of protein sequences and biomedical texts. In Proc. 40th International Conference on Machine Learning (eds Krause, A. et al.) 38749–38767 (PMLR, 2023).
Chen, J. et al. Global marine microbial diversity and its potential in bioprospecting. Nature 633, 371–379 (2024).
Hu, Z. et al. Discovery and engineering of small SlugCas9 with broad targeting range and high specificity and activity. Nucleic Acids Res. 49, 4008–4019 (2021).
Yu, T. et al. Enzyme function prediction using contrastive learning. Science 379, 1358–1363 (2023).
Kweon, J. et al. Efficient DNA base editing via an optimized DYW-like deaminase. Preprint at bioRxiv https://doi.org/10.1101/2024.05.15.594452 (2024).
Gherardini, P. F., Wass, M. N., Helmer-Citterich, M. & Sternberg, M. J. E. Convergent evolution of enzyme active sites is not a rare phenomenon. J. Mol. Biol. 372, 817–845 (2007).
Doolittle, R. F. Convergent evolution: the need to be explicit. Trends Biochem. Sci. 19, 15–18 (1994).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Pomaznoy, M., Ha, B. & Peters, B. GOnet: a tool for interactive Gene Ontology analysis. BMC Bioinformatics 19, 470 (2018).
Dauparas, J. et al. Robust deep learning–based protein sequence design using ProteinMPNN. Science 378, 49–56 (2022).
He, Y. et al. Protein language models-assisted optimization of a uracil-N-glycosylase variant enables programmable T-to-G and T-to-C base editing. Mol. Cell 84, 1257–1270 (2024).
Tong, H. et al. Development of deaminase-free T-to-S base editor and C-to-G base editor by engineered human uracil DNA glycosylase. Nat. Commun. 15, 4897 (2024).
Ye, L. et al. Glycosylase-based base editors for efficient T-to-G and C-to-G editing in mammalian cells. Nat. Biotechnol. 42, 1538–1547 (2024).
Cornman, A. et al. The OMG dataset: an Open MetaGenomic corpus for mixed-modality genomic language modeling. In Proc. 13th International Conference on Learning Representations (ICLR, 2025); https://openreview.net/forum?id=jlzNb1iWs3
Kavli, B. et al. Excision of cytosine and thymine from DNA by mutants of human uracil-DNA glycosylase. EMBO J. 15, 3442–3447 (1996).
Hayes, T. et al. Simulating 500 million years of evolution with a language model. Science 387, 850–858 (2025).
Burley, S. K. et al. RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 47, D464–D474 (2019).
Richardson, L. et al. MGnify: the microbiome sequence data analysis resource in 2023. Nucleic Acids Res. 51, D753–D759 (2023).
Pruitt, K. D., Tatusova, T., Brown, G. R. & Maglott, D. R. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 40, D130–D135 (2012).
Dai, F. et al. Toward de novo protein design from natural language. Preprint at bioRxiv https://doi.org/10.1101/2024.08.01.606258 (2024).
Liu, N. et al. Protein design with dynamic protein vocabulary. Preprint at https://arxiv.org/abs/2505.18966 (2025).
Kuang, J., Liu, N., Sun, C., Ji, T. & Wu, Y. PDFBench: a benchmark for de novo protein design from function. Preprint at https://arxiv.org/abs/2505.20346 (2025).
Ko, Young Su. Using ProTrek for protein binder design. Twitter https://x.com/youngsuko9/status/1865845977673834595 (2024).
Gitter, A. Using ProTrek to retrieve proteins with desired function. Twitter https://x.com/anthonygitter/status/1827760237194920435 (2024).
Gitter, A. Using ProTrek to retrieve proteins with desired function. Twitter https://x.com/anthonygitter/status/1813427191000035330 (2024).
Gitter, A. Using ProTrek to retrieve proteins with desired function. Twitter https://x.com/anthonygitter/status/1882642214624678193 (2025).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
van den Oord, A., Li, Y. & Vinyals, O. Representation learning with contrastive predictive coding. Preprint at https://arxiv.org/abs/1807.03748 (2018).
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. BERT: pre-training of deep bidirectional transformers for language understanding. In Proc. 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 1 (Long and Short Papers) 4171–4186 (Association for Computational Linguistics, 2019).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Rasley, J., Rajbhandari, S., Ruwase, O. & He, Y. DeepSpeed: system optimizations enable training deep learning models with over 100 billion parameters. In Proc. 26th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (eds Gupta, R. & Liu, Y.) 3505–3506 (Association for Computing Machinery, 2020).
Loshchilov, I. and Hutter, F. Fixing weight decay regularization in Adam. OpenReview.net https://openreview.net/forum?id=rk6qdGgCZ (2018).
Loshchilov, I. & Hutter, F. SGDR: Stochastic gradient descent with warm restarts. In Proc. International Conference on Learning Representations (ICLR, 2017); https://openreview.net/forum?id=Skq89Scxx
Xu, J. et al. Protein inverse folding from structure feedback. Preprint at https://arxiv.org/abs/2506.03028 (2025).
Enzyme Nomenclature (Nomenclature Committee of the International Union of Biochemistry and Molecular Biology, 2024); https://iubmb.qmul.ac.uk/enzyme/
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361 (2017).
Kucera, T., Oliver, C., Chen, D., and Borgwardt, K. ProteinShake: building datasets and benchmarks for deep learning on protein structures. In Advances in Neural Information Processing Systems 36 (eds Oh, A. et al.) (NeurIPS, 2023).
Acknowledgements
We thank P. Beltrao, S. Ovchinnikov, J. Zeng, C. Wang and J. Huang for their valuable suggestions to improve this paper. We thank L. Hong and M. Li for providing the predownloaded OMG and MGnify databases. This work is supported by Zhejiang Province Leading Geese Plan (2025C01094), the National Natural Science Foundation of China (32471547, U21A20427, 82450102 and 32025016), the Ministry of Science and Technology of the People’s Republic of China (2022YFA0807300), the National Key Research and Development Program of China (2022ZD0115100), the Zhejiang Key Laboratory of Low-Carbon Intelligent Synthetic Biology (2024ZY01025), the Guangzhou Science and Technology Program City–University Joint Funding Project (2023A03J0001), the Nansha Key Science and Technology Project (2023ZD015), the Guangdong Education Department (2023ZDZX2073), the Hong Kong University of Science and Technology 20 for 20 Cross-Campus Collaborative Research Scheme (G051) and the Westlake Center of Synthetic Biology and Integrated Bioengineering. We also thank N. Li and the Westlake HPC Center for computing resources and technical support.
Author information
Authors and Affiliations
Contributions
F.Y. conceptualized and led this research. J.S., Y.H. and S.Y. performed the main research. H.L. and X.C. supervised and led the wet-lab experiments and analyses. J.S. and F.Y. designed the ProTrek network architecture. J.S. conducted the machine learning modeling and implementation. J.S. and X. Zhou collected and curated the training datasets. J.S., S.J. and I.T. performed the computational experimental analyses. I.T., H.L. and X.C. provided bioinformatics guidance. X. Zhang developed the ColabProTrek. Y.H., S.Y., X.S., H.L. and X.C. conducted the wet-lab experiments and evaluations. Y.W. explored an early wet-lab validation experiment. F.Y., J.S., Y.H., X.C. and H.L. wrote and revised the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Biotechnology thanks Wei Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Evaluation of Hit Rates for Enzyme Commission (EC) Numbers and CRISPR-associated Proteins Using ProTrek’s Text-to-Sequence Retrieval Function.
Textual descriptions were used to query the UniProt50 database and extract the top 100 proteins not included in the training data. a, Bar plot depicts hit rate for six EC numbers selected from six top-level EC classes, categorized into exact matches and varying levels of mismatches (Mismatch-1: mismatch in the last digit; Mismatch-2: mismatch in the last two digits; Mismatch-3: only match the first digit; Mismatch: mismatch all digits). Each bar shows the proportion of exact matches and progressively less accurate matches. The results demonstrate ProTrek’s performance in identifying enzyme categories, with variations in match accuracy across different EC numbers. b, Bar plot illustrates hit rate for six extensively studied CRISPR-associated proteins (Cas10, Cas3, Cas9, Cas12, Cas13, and Csm/Cmr). Each bar is segmented into matches and mismatches, showcasing ProTrek’s retrieval performance across these protein categories. If the corresponding keywords such as ‘Cas10’, ‘Cas3’, ‘Cas9’, ‘Cas12’, or ‘EC:3.4.15.1’ appear in the protein’s ground truth, it is considered a true hit.
Extended Data Fig. 2 Structural similarity matrix of the top 30 exact matched enzymes.
The matrix shows the pair-wise TM-scores for the top 30 exact matched enzymes that were shown in Extended Data Fig. 1a. The value of each cell in the matrix denotes the TM-score, indicating the degree of structural similarity for each protein pair. The color scale indicates the TM-score value, where warmer colors represent higher structural similarity and cooler colors represent lower similarity.
Extended Data Fig. 3 Structural similarity matrix of the top 30 identified CRISPR-associated proteins.
The matrix displays the pair-wise TM-scores for the top 30 matched CRISPR-associated proteins that were shown in Extended Data Fig. 1b. The value of each cell in the matrix denotes the TM-score, indicating the degree of structural similarity for each protein pair. The color scale indicates the TM-score value, where warmer colors represent higher structural similarity and cooler colors represent lower similarity.
Extended Data Fig. 4 Experimental validation of proteins identified through ProTrek searches.
a, Sequence alignment of hUDG-Y147A with proteins identified via ProTrek searches. S-V1 to S-V5 represent the top five hits obtained from the sequence-to-sequence search, while T-V1 to T-V5 represent the top five hits obtained from the text-to-sequence search. Yellow-highlighted regions denote highly conserved sequences. Sequence alignment was performed using Clustal Omega. b, Proteins S-V1 to S-V5 and T-V1 to T-V5 were fused with Cas9n following the introduction of a mutation analogous to UDG-Y147A. Notably, S-V1 and T-V1 refer to the same protein. eGFP was used as a negative control, while the mock group represented cells without any treatment. HeLa cells were transfected with the base editor constructs alongside specific sgRNAs targeting Dicer-1 and VEGFA. Five days post-transfection, thymine nucleotide substitutions at the target sites were quantified using high-throughput sequencing (HTS), with the mutated nucleotide positions annotated relative to the 5’ end of the protospacer. Data are presented as mean ± s.d. from two independent experiments (n = 2).
Extended Data Fig. 5 Alignment speed comparison (CPU time) for 100 query proteins against the UniRef50 database, using 24 CPU cores.
ProTrek demonstrates efficient database processing and searching capabilities with rapid response times, achieving over 100-fold speed improvement compared to Foldseek and MMseqs2.
Extended Data Fig. 6 ProTrek score sensitivity analysis for keyword and template regions in functional descriptions.
We first computed the similarity score (‘Score_original’) between a protein sequence and its complete textual description. Next, we systematically removed each word from the description and recalculated the similarity score with the protein. For the template region (blue circle), we averaged all similarity scores obtained after deleting words within this region to derive ‘Score_changed’. Similarly, for the keyword region (orange triangle), ‘Score_changed’ was calculated by averaging all similarity scores resulting from the removal of words in that specific region.
Supplementary information
Supplementary Information (download PDF )
Supplementary Tables 1–16 and Figs. 1–9.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Su, J., He, Y., You, S. et al. A trimodal protein language model enables advanced protein searches. Nat Biotechnol (2025). https://doi.org/10.1038/s41587-025-02836-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41587-025-02836-0
This article is cited by
-
Learning physical interactions to compose biological large language models
Communications Chemistry (2026)
-
Ab-initio amino acid sequence design from protein text description with ProtDAT
Nature Communications (2025)
-
Democratizing protein language model training, sharing and collaboration
Nature Biotechnology (2025)
