
Abstract
CRISPR–Cas3 represents a mechanistically distinct genome-editing system compared to Cas9 that generates long-range deletions rather than small indels, thereby reducing the risk of residual protein function from in-frame mutations. Here we evaluated CRISPR–Cas3 to correct mutations in the TTR gene causing transthyretin amyloidosis, a systemic proteinopathy where loss of mutant TTR in the liver offers therapeutic benefit. Through CRISPR RNA optimization we achieved 58.9% ± 0.5% editing at the TTR locus in vitro, inducing large deletions that abolished TTR expression. Cas3 generated mostly directional deletions up to 75 kb without reproducible off-target mutations, in contrast to Cas9, which induced indels at several off-target sites. In vivo, a single lipid-nanoparticle-based treatment achieved 48.7% ± 1.1% hepatic editing and reduced serum TTR levels by 80.1% ± 4.6%. Deletion size was limited to 21 kb. In TTR exon-humanized mice, Cas3 editing reduced serum TTR without in-frame mutations and attenuated macrophage-associated TTR deposition. These findings highlight Cas3 as an efficient and distinct sytem for in vivo genome editing.
Similar content being viewed by others

Main
Transthyretin amyloidosis (ATTR) is a systemic protein misfolding disorder caused by the deposition of amyloid fibrils derived from transthyretin (TTR), a tetrameric protein primarily synthesized in the liver1,2,3. ATTR exists in two major forms: hereditary (ATTRv), resulting from destabilizing mutations in the TTR gene4,5, and wild-type (ATTRwt), which is age associated and more prevalent. ATTRwt affects an estimated 500,000 individuals worldwide, with TTR deposition identified in approximately 25% of autopsied hearts in individuals over 80 years old6,7,8,9. Current therapeutic options include TTR stabilizers, which prevent tetramer dissociation and aggregation, and nucleic-acid-based agents such as small interfering RNA and antisense oligonucleotides that reduce hepatic TTR synthesis1,10,11,12,13. While these therapies effectively lower circulating TTR and slow disease progression, they do not address the underlying genetic defect and typically require repeated, lifelong administration.
Genome-editing strategies, particularly those based on CRISPR–Cas9, have recently entered clinical development14,15. A notable example is NTLA-2001, a lipid nanoparticle (LNP)-delivered mRNA–Cas9 therapeutic that targets TTR in vivo. In persons with hereditary ATTR, NTLA-2001 has demonstrated durable reductions in serum TTR and is now in phase 3 clinical trials16,17. Moreover, a long-term follow-up reported up to 90% sustained plasma TTR reduction over 24 months, further supporting the clinical promise of in vivo CRISPR therapeutics18. In parallel, other in vivo gene-editing programs using Cas9, base editors or prime editors have entered early-phase trials for other monogenic disorders and viral infections16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36 (Supplementary Table 1). Cas9 editing induces double-strand breaks (DSBs) that are repaired through nonhomologous end joining (NHEJ), which typically results in small insertions or deletions (indels)37 or rare large deletions or chromosomal rearrangements38. Depending on the indel pattern, the reading frame may be preserved, potentially resulting in in-frame mutations (IFMs) that may yield aberrant protein products.
CRISPR–Cas3, a class 1 type I-E system, features a multisubunit Cascade complex (Cas5, Cas6, Cas7, Cas8, Cas11 and CRISPR RNA (crRNA)) that recognizes target DNA and recruits the Cas3 helicase–nuclease, enabling processive and unidirectional DNA degradation39,40,41,42,43. This contrasts with the localized DSB introduced by class 2 systems like Cas9. Cas3 typically produces long deletions upstream of the protospacer-adjacent motif (PAM), a property that may reduce the likelihood of generating short IFMs that could preserve residual gene function. In addition to DNA cleavage, inactivated Cascade complexes have been adapted for transcriptional and epigenetic regulation in human and plant cells44,45,46. Cas3 has also been used in vivo to induce large deletions in animal models, including mice47, supporting its feasibility for genome editing in mammals.
In this study, we evaluated CRISPR–Cas3 for genome editing of the TTR gene. We examined its efficiency and specificity in mouse Hepa1-6 cells, in vivo mouse liver and human HepG2 cells using both plasmid and/or mRNA–LNP delivery. Off-target effects were comprehensively analyzed using whole-genome sequencing (WGS) with both short-read and long-read technologies. In human cells, Cas3 editing outcomes were compared to Cas9 using the clinically validated NTLA-2001. We also analyzed in vivo effects, including serum TTR reduction and cardiac deposition in TTR exon-humanized mice. Although Cas9-based therapeutics such as NTLA-2001 remain the clinical benchmark for ATTR, our study provides an initial evaluation of Cas3 as a distinct and complementary genome-editing approach.
Results
Screening of crRNAs for Ttr gene knockout in Hepa1-6 cells
To identify effective crRNAs for CRISPR–Cas3-mediated editing of the mouse Ttr gene, we designed five crRNA candidates (crRNA1–crRNA5) targeting exons 1 and 2 (Fig. 1a, Supplementary Fig. 1 and Supplementary Table 2). Each crRNA was cloned into an all-in-one plasmid (pRB-EF1α-Cas3-Cascade-U6v2-BbsI-GFP, referred to as pCas3), which coexpresses a mammalian codon-optimized Cascade–Cas3 complex and EGFP for transfection enrichment (Extended Data Fig. 1 and Supplementary Data 1). Mouse hepatoma Hepa1-6 cells were transfected with pCas3 constructs and EGFP-positive cells were sorted by fluorescence-activated cell sorting (FACS; BD FACSAria, BD Biosciences). Genomic DNA (gDNA) was extracted and subjected to PCR and agarose gel electrophoresis. The appearance of smaller amplicons indicated Cas3-induced genomic deletions at the targeted loci (Fig. 1b). Among the candidates, crRNA2, which exhibited the highest editing activity with a droplet digital PCR (ddPCR)-measured deletion efficiency of 58.9% ± 0.5% in exon 2 (Fig. 1c), was used for subsequent off-target and in vivo experiments.

a, Schematic of the mouse Ttr (ENSMUST00000075312.5) and B4galt6 (ENST00000306851) loci, showing five Cas3-targeting crRNAs (crRNA1–crRNA5) and one Cas9 gRNA (G211). Primer pairs used for conventional PCR amplification are indicated, along with the genomic regions interrogated by ddPCR probes for quantitative assessment. b, Analysis of genome-editing efficiency in Hepa1-6 cells following transfection with pCas3-crRNA or px458-gRNA (for Cas9). PCR amplicons of the Ttr target region were visualized by agarose gel electrophoresis. Appearance of smaller amplicons in Cas3-transfected samples confirms efficient cleavage at the intended target sites. NC, nontransfected control. c, ddPCR-based quantification of deletion efficiency at Ttr exon 2. Transfection with pCas3-cr2 yielded ~60% deletion, significantly higher than other crRNAs. Statistical analysis was performed using a one-way ANOVA with Dunnett’s post hoc test versus pCas3-cr2 (n = 3 independent biological replicates per group (separately seeded and transfected Hepa1-6 cell cultures)). d, ddPCR-based quantification of editing at Ttr exon 4 and off-target effects at B4galt6 exon 9 in pCas3-cr2-transfected cells. A deletion frequency of 30.9% ± 2.2% was detected at Ttr exon 4, while off-target editing at B4galt6 exon 9 remained at 4.4% ± 2.0%. No detectable deletions were observed in px458-G211-treated cells. Statistical test was performed using a one-way ANOVA with Dunnett’s post hoc test (versus NC). Error bars indicate the mean ± s.e.m. (n = 3 independent biological replicates per group (separately seeded and transfected Hepa1-6 cell cultures)).
Evaluation of on-target and potential off-target mutations
Given the processive exonuclease activity of CRISPR–Cas3, which enables extended DNA degradation39,43, we evaluated the genomic editing range in Hepa1-6 cells using ddPCR to quantify potential downstream effects. We analyzed exon 4 of the Ttr gene and exon 9 of the neighboring β-GlcNAc β1,4-galactosyltransferase, polypeptide 6 (B4galt6) gene, located downstream of Ttr (Fig. 1a). In cells edited with Cas3 and crRNA2, 30.9% ± 2.2% of Ttr exon 4 alleles showed deletions, while B4galt6 exon 9 exhibited minimal editing (4.4% ± 2.0%) (Fig. 1d). In contrast, Cas9 editing (PX458; Addgene, 48138)48 using the G211 single guide RNA (sgRNA)49 produced no detectable deletions at either locus. These results suggest that Cas3-mediated deletions can extend several kilobases upstream of the PAM39,43.
To further characterize the deletion profile, we performed targeted capture sequencing (CapSeq) using probes spanning a 100-kb window around the Ttr locus (90 kb upstream and 10 kb downstream of the PAM). This high-resolution approach revealed a clear peak of split reads corresponding to Cas3-induced deletions (Fig. 2a). Among 1,337 edited reads, deletion lengths ranged from 25 bp to 74,709 bp, with a mean of 8,696 bp. In a subset of reads (117/1,337), deletions extended into the final exon or 3′ untranslated region (UTR) of B4galt6. Deletion initiation sites clustered within 200 bp upstream of the PAM-proximal cascade-binding site (Extended Data Fig. 2a), consistent with our previous biochemical analyses39,50. However, termination points of the deletions appeared more variable, without enrichment for specific sequence motifs. These findings are consistent with Cas3’s characteristic unidirectional, long-range deletion activity49

a, CapSeq of the Ttr locus in pCas3-cr2-treated Hepa1-6 cells. Capture probes spanned a 100-kb region (from 90 kb upstream to 10 kb downstream of the PAM). Among 1,337 reads analyzed, deletions occurred predominantly upstream (88%), with an average size of 9,192 bp and a maximum of 74,709 bp, whereas downstream deletions (12%) averaged 5,160 bp with a maximum of 14,759 bp. b, The number of POT sites identified for Cas3 crRNA2 and Cas9 gRNA (G211) using GGGenome and Cas-OFFinder. For POT sites, capture probes were designed to cover 5 kb around Cas3 sites and 2 kb around Cas9 sites. c, CapSeq showed weak signals at Cas3 POT93 and POT295, likely because of alignment artifacts in repetitive regions (Extended Data Fig. 3). d, Cas9-treated samples exhibited distinct off-target indels at POT1 and POT129. Representative aligned read sequences at these sites are shown with corresponding percentages and read counts.
We next predicted genome-wide potential off-target (POT) sites using GGGenome (https://GGGenome.dbcls.jp/) and Cas-OFFinder51, applying search parameters based on the 32-nt 5′ segment of crRNA2 (excluding every sixth base, which does not engage in target recognition51). This analysis identified 189 candidate sites with partial sequence identity (16–19-nt matches) and 166 sites with ≤7 mismatches. For Cas9 G211, 653 POT sites with ≤5 mismatches were identified under comparable conditions (Fig. 2b and Supplementary Tables 3 and 4). To assess whether Cas3 or Cas9 induced detectable off-target mutations at these sites, we performed high-depth CapSeq using focused probe windows: 5 kb for Cas3 POT sites and 2 kb for Cas9 POT sites. In Cas3-edited samples, weak signal enrichment was observed at POT93 and POT295 (Fig. 2c) but manual inspection of the aligned reads and RepeatMasker tracks indicated that these were located within repetitive elements and likely represented mapping artifacts (Extended Data Fig. 2b). By contrast, Cas9-treated samples showed reproducible off-target indels at POT1 and POT129, which were confined to intergenic regions (Fig. 2d).
We further evaluated genome-wide specificity using both short-read (139.5×) (Extended Data Fig. 3a,b) and Oxford Nanopore long-read (28.4×) WGS (Extended Data Fig. 3c,d). In short-read data, 1,037 candidate deletion regions with split-read ratios ≥ 1.38 (corresponding to >10% estimated editing) were detected in Cas3-treated cells. However, all candidate regions outside the Ttr locus corresponded to repetitive or low-complexity genomic regions (Extended Data Fig. 3e,f). Long-read WGS similarly revealed a single major peak at the on-target Ttr locus, with no prominent peaks detected at POT sites or elsewhere in the genome (Extended Data Fig. 3g,h). Although sporadic mutations were detected genome-wide in both Cas3-treated and control samples, these likely reflect background artifacts because of relaxed detection thresholds for rare nongermline deletions.
Optimization of mRNA modifications and constructs for efficient genome editing
To improve mRNA stability and translation while minimizing immunogenicity, we applied nucleotide modifications commonly used in therapeutic mRNA delivery52,53. In Hepa1-6 cells transfected with GFP mRNA, we tested various combinations of internal nucleotide modifications and 5′ cap structures. Regardless of the presence of N1-methylpseudouridine (N1mΨ), the Cap1 structure consistently enhanced GFP fluorescence without affecting cell viability (Extended Data Fig. 4a,b). On the basis of these results, we adopted similar modifications for all six components of the CRISPR–Cas3 system, synthesizing each mRNA with or without N1mΨ and Cap1.
To evaluate the optimal ratio between Cas3 mRNA and crRNA, we tested three ratios in Hepa1-6 cells: a baseline equal-mass mix, a fourfold excess of crRNA and a twofold increase in both Cas3 and crRNA (Extended Data Fig. 4c). Editing outcomes appeared comparable across conditions in this exploratory assay, suggesting that Cas3-mediated editing may tolerate moderate variation in component ratios. However, a comprehensive analysis of stoichiometric dependencies among all six components remains to be conducted.
We also evaluated three different mRNA delivery strategies: all-in-one, encoding all six Cas genes in a single transcript, Separated (Sep), where each gene was delivered on an individual mRNA, and divide-in-two (DiT), grouping Cas3, Cas5 and Cas11 in one mRNA and Cas6, Cas7, and Cas8 in another (Extended Data Fig. 4d). All constructs incorporated nuclear localization signals (NLSs) and 2A self-cleaving peptides to ensure proper localization and efficient translation. While statistical differences were not significant, ddPCR analysis showed that Sep and DiT configurations yielded higher editing efficiency when used with Cap1 and N1mΨ-modified mRNAs (Extended Data Fig. 4e).
To further enhance editing, we screened five chemically modified versions of crRNA2 targeting the Ttr gene (Extended Data Fig. 4f). The most effective design, crRNA2 Stem1 ver2, included 2′-O-methylation54 and phosphorothioate bonds55 at both termini and the stem-loop region, which improved RNA stability and reduced nuclease degradation. This optimized crRNA yielded the highest genome-editing efficiency in Hepa1-6 cells (Extended Data Fig. 4g). These results suggest that fine-tuning of mRNA and crRNA modifications improved the editing efficiency in vitro.
Dose-dependent in vivo Cas3 editing in ICR mice
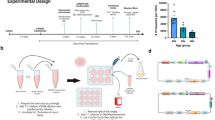
To evaluate the genome-editing capability of CRISPR–Cas3 in vivo, we intravenously administered LNPs containing chemically modified crRNA (TTR2 Stem1 ver2) and Cascade–Cas3 mRNAs, formulated in either Sep or DiT configurations, to 1-month-old Jcl:ICR mice. Genome-editing efficacy was assessed by measuring serum TTR levels using ELISA at 1, 2, 3 and 4 weeks after injection. Then, 1 week after administration, serum TTR levels were reduced by 71.3% ± 7.1% in the Sep-Cas3–LNP group and 77.7 ± 4.1% in the DiT-Cas3–LNP group relative to saline-treated controls (Fig. 3a). This reduction was dose dependent and sustained for several weeks (Fig. 3b).

a, Serum TTR levels measured 1 week after administration of LNPs or saline. At a dose of 6 mg kg−1, Sep-Cas3–LNP and DiT-Cas3–LNP reduced TTR levels by approximately 71% and 77%, respectively, demonstrating a dose-dependent effect compared to lower doses and saline controls (n = 3–4 biological replicates per group). b, Time course of serum TTR levels in ICR mice treated with saline, Sep-Cas3–LNP or DiT-Cas3–LNP (6 mg kg−1; n = 3 biological replicates per group). TTR levels remained low, indicating a durable effect of genome editing. c, TTR protein expression was evaluated by immunohistochemistry in liver sections from ICR mice 4 weeks after treatment with DiT-Cas3–LNP (6 mg kg−1) or saline. A marked reduction in TTR staining (brown, DAB) was observed in hepatocytes of Cas3-treated mice. Nuclei were counterstained with DAPI (blue). d, ddPCR analysis of liver gDNA from ICR mice 4 weeks after DiT-Cas3–LNP treatment (6 mg kg−1) revealed ~50% deletion at Ttr exon 2 (n = 3 biological replicates per group). e, CapSeq of the Ttr locus in hepatocytes from DiT-Cas3–LNP-treated mice identified Cas3-induced deletions. Among 3,178 reads analyzed, deletions occurred predominantly upstream (84%) with an average size of 3,304 bp and a maximum of 20,506 bp, whereas downstream deletions (16%) averaged 4,085 bp with a maximum of 14,573 bp. Deletions extending into the neighboring B4galt6 gene were confined to its 3′ UTR, with no exonic disruption detected. f, Amplicon sequencing of liver DNA from mice treated with Cas9/G211–LNP (2 mg kg−1) showed off-target indel mutations at predicted sites POT1 and POT129, corroborating the CapSeq findings in vitro (Fig. 2d). Error bars represent the mean ± s.e.m. Statistical analysis was conducted using a two-tailed Student’s t-test versus saline (a) or a two-tailed Welch’s t-test (d).
To further improve editing efficiency, we introduced PureCap Cap2 modifications into the Cascade–Cas3 mRNAs. Cap2 has been shown to enhance translation efficiency by 3–4-fold compared to Cap1 in both cultured cells and animal models56. N1mΨ-modified mRNAs for Cascade–Cas3 mRNAs were synthesized in-house using a Cap2 cap analog (TetraPure CAG)56 and formulated into LNPs (Sep-Cas3-Cap2). Administration of Sep-Cas3-Cap2–LNPs resulted in 80.1% ± 4.6% reduction in serum TTR, indicating enhanced genome-editing activity in vivo (Extended Data Fig. 5a). Reverse transcription (RT)–qPCR confirmed that Ttr mRNA levels in the liver were reduced by ~86% following administration of Sep-Cas3–LNP or DiT-Cas3–LNP (6 mg kg−1) (Extended Data Fig. 5b). Immunohistochemical staining further confirmed effective TTR protein knockdown in the liver (Fig. 3c).
Then, 4 weeks after DiT-Cas3–LNP administration (6 mg kg−1), ddPCR of liver gDNA indicated an editing efficiency of 48.7% ± 1.1% (Fig. 3d). Compared to plasmid-based delivery, mRNA–LNP delivery allows a transient expression window, which reduces the risk of off-target effects and improves the safety profile. CapSeq of the Ttr locus in hepatocytes from DiT-Cas3–LNP-treated mice revealed narrower deletion distributions than those observed in pCas3-treated Hepa1-6 cells (Fig. 2a). Deletion initiation sites clustered again within ~200 bp upstream of the PAM-proximal Cascade-binding site (Extended Data Fig. 2c). In vivo, 84% of deletions occurred upstream of the protospacer and 16% occurred downstream, extending up to 20.5 kb (Fig. 3e). Among 3,178 split reads, only 18 extended into the neighboring B4galt6 gene and these were restricted to its 3′ UTR, with no deletions detected in exon 9. In LNP-mediated Cas3 editing, deletion distributions were narrower than with plasmid-based delivery. In contrast, amplicon sequencing of livers from Cas9/G211–LNP–treated mice revealed clear off-target editing at POT1 and POT129 (Fig. 3f), both in noncoding regions, consistent with in vitro observations (Fig. 2d). Cas3-mediated editing was not detected in other organs such as the spleen or kidney (Extended Data Fig. 5c), supporting the tissue-specific activity of the delivered LNPs. No overt toxicity signals were observed during the 3–6-month follow-up period.
Immunogenicity and tolerance of Cas3–LNPs
LNP-based delivery systems have raised safety concerns because of potential immunogenicity and hepatic accumulation. To evaluate these risks, we monitored serum cytokine levels (interferon-α (IFNα), tumor necrosis factor (TNF), interleukin 6 (IL-6) and monocyte chemoattractant protein 1 (MCP1)) at 4, 24 and 72 h, as well as at 1 and 2 weeks after DiT-Cas3–LNP administration, following a protocol similar to that used in Cas9–LNP studies49. Cytokine levels transiently increased at 4 h after injection but returned to baseline within 1 week, similar to those observed in the empty LNP (formulated without mRNA) and saline control groups (Fig. 4a). Although direct comparisons to Cas9–LNP are limited by differences in dosing (0.3–2 mg kg−1 for Cas9 (ref. 49) versus 6 mg kg−1 for Cas3), the temporal pattern of cytokine changes appeared similar across groups. Nonetheless, these findings do not constitute a full toxicological or safety assessment and long-term studies will be required to evaluate immunogenicity, tolerability and delayed pathological changes.

a, Serum cytokine levels (IFNα, TNF, IL-6 and MCP1) were measured at 4, 24, 72, 168 and 336 h following intravenous administration of DiT-Cas3–LNP (6 mg kg−1), empty LNP or saline. Both DiT-Cas3–LNP and empty LNP induced a transient increase in cytokine levels at 4 h compared to saline. These responses declined by 72 h, with a reduction of 70–85%, and returned to baseline by 168–336 h. Error bars represent the mean ± s.e.m. (n = 3–5 biological replicates per group). b, Western blot analysis of liver homogenates collected at 4, 6, 24 and 168 h after treatment with DiT-Cas3–LNP (6 mg kg−1) or saline. Cas3 protein was detectable at 4 and 6 h, diminished at 24 h and undetectable at 168 h, indicating transient protein expression. Recombinant Cas3 protein was used as a positive control; α-tubulin served as a loading control.
To investigate the kinetics of Cas3 activity in vivo, PCR amplification of the target region confirmed the onset of genome editing within 4 h after injection (Extended Data Fig. 5d). Western blot analysis detected Cas3 protein in the liver at 4 and 6 h, with levels decreasing by 24 h and becoming undetectable by 168 h (Fig. 4b), indicating rapid clearance. RT–qPCR further showed that Cascade RNA transcripts were degraded within 1 week (Extended Data Fig. 5e). The rapid clearance of both protein and RNA components likely reduces the risk of prolonged or unintended off-target activity, consistent with transient, liver-restricted activity under these conditions.
Cas3 in vivo editing in humanized mouse models
To assess genome editing of the human TTR gene in vivo, we used a TTR exon-humanized mouse model in which all mouse Ttr coding exons were replaced with their human counterparts, while intronic and regulatory elements remain murine in origin57 (Fig. 5a). This model recapitulates human TTR expression and has been used to study ATTRwt. We tested whether crRNA2—originally optimized for the mouse Ttr sequence—would also reduce serum human TTR levels. The Cas9 sgRNA NTLA-2001, currently in phase 3 clinical trials16,17, was used as a reference.

a, Schematic representation of the humanized TTR gene structure in exon-humanized mice. The locations of the Cas3 crRNA2, Cas9 gRNA NTLA-2001 and the ddPCR probe are indicated. b, Label-free quantification (LFQ) of the mutant TTR peptide (AADAWEPFASGK), corresponding to IFM4, in serum from untreated (n = 1 per group), Cas3-treated (DiT-Cas3–LNP, 6 mg kg−1; n = 3 biological replicates per group) and Cas9-treated (NTLA-2001, 2 mg kg−1; n = 3 biological replicates per group) mice. The mutant peptide was detected only in the Cas9 group by nanoLC–MS/MS, suggesting the formation of edited transcripts with an altered amino acid sequence. c, Serum TTR levels measured 1 week after Cas3 treatment (3.8–4.6 mg kg−1, n = 5 biological replicates per group) in 8-month-old exon-humanized mice showed a reduction of 77.8% ± 21.1% compared to untreated controls (n = 3 biological replicates per group). d, Immunofluorescence analysis of cardiac tissue from exon-humanized mice at 2 months after treatment. Representative confocal images showing TTR (green), CD68-positive macrophages (red) and DAPI-stained nuclei (blue). e, Quantification of DAPI-positive nuclei, TTR-positive foci and CD68-positive foci. f, Quantification of TTR-positive area and CD68-colocalized TTR signals (CD68/TTR ratio), normalized to untreated controls (n = 5 biological replicates per group). Cas3 treatment (n = 5 biological replicates per group) resulted in reduced cardiac TTR deposition and attenuated macrophage accumulation at deposition sites. Error bars represent the mean ± s.e.m. Statistical analysis was conducted using a two-tailed Student’s t-test for serum TTR (c), the number of DAPI per field (e) and the number of CD68 foci (f) or a two-tailed Welch’s t-test for the number of TTR foci (e).
The 1-month-old exon-humanized mice received a single tail-vein injection of DiT-Cas3–LNP or Cas9/NTLA-2001–LNP. Then, 1 week after administration, serum human TTR levels were significantly reduced in both groups: by 74.6% ± 0.6% in the DiT-Cas3–LNP group and by 94.9% ± 0.3% in the Cas9/NTLA-2001–LNP group (Extended Data Fig. 6a). Using sensitive nano liquid chromatography–tandem mass spectrometry (nanoLC–MS/MS) analysis58, we detected a mutant TTR peptide (AADAWEPFASGK, IFM4; p.D39_40TdelinsA) in the serum of Cas9-treated mice but not in Cas3-treated mice (Fig. 5b and Extended Data Fig. 6b). AlphaFold2 predictions59,60 indicated that IFM4 alters local β-sheet structures in the TTR monomer (Extended Data Fig. 6c–g). In vitro assays showed that the recombinant IFM4 peptide had increased aggregation under acidic conditions (pH 4.5), similar to the well-characterized V30M variant in ATTRv61 (Extended Data Fig. 6h). SDS–PAGE revealed that IFM4 and V30M failed to form tetramers, unlike WT TTR (Extended Data Fig. 6i). Amplicon sequencing of Cas9/NTLA-2001-edited mouse embryonic fibroblasts (MEFs) derived from the humanized model (Extended Data Fig. 7a) revealed various IFMs, with IFM4 comprising ~10% of edited alleles (Extended Data Fig. 7b,c), whereas Cas3 editing did not generate IFMs in our analysis.
Consistent with other mouse models of ATTR62,63, our exon-humanized mice showed no overt cardiac TTR amyloid deposition detectable by DFS staining at 9 months of age (Extended Data Fig. 8a). However, TTR deposition with CD68⁺ macrophage accumulation was observed by fluorescence immunostaining, starting around 9 months of age and increasing from 2 months (Extended Data Fig. 8b–d), consistent with early-stage ATTR pathology reported in both humans64,65 and mice63. To evaluate whether Cas3 editing could modulate this phenotype, we administered DiT-Cas3–LNP to 8-month-old humanized mice. Here, 1 week after treatment, serum TTR levels were reduced by 77.8% ± 9.4% (Fig. 5c). Then, 2 months after injection, immunofluorescence analyses revealed a marked reduction in cardiac TTR deposition and associated macrophage infiltration compared to untreated aged controls (Fig. 5d–f and Extended Data Fig. 8e,f). No overt toxicity or sudden death was observed in Cas3-treated mice during the observation period.
In vitro assessment of Cas3 targeting at the human TTR locus
We further conducted a comprehensive screening of candidate crRNAs in HepG2 cells, a human hepatocyte-derived cell line. Ten crRNAs targeting the human TTR gene (UT01–UT10; Fig. 6a, Supplementary Fig. 2 and Supplementary Table 5) were cotransfected with an all-in-one pCas3 plasmid (Extended Data Fig. 1) and editing efficiency was assessed by ddPCR and RT–qPCR. The clinically validated Cas9 sgRNA NTLA-2001 (refs. 16,17) was used as an established reference under matched conditions. In ddPCR assays targeting TTR exon 2, crRNA UT05 demonstrated editing efficiency that was not significantly different from NTLA-2001 (P = 0.0777) (Fig. 6b). RT–qPCR analyses using primer sets spanning exons 1–2 and exons 3–4 further identified multiple crRNAs—including UT05, UT06 and UT07— as yielding comparable levels of TTR mRNA reduction (P > 0.05) (Fig. 6c).

a, Schematic representation of the human TTR gene (ENST00000237014.8), indicating the positions of the candidate crRNAs (UT01–UT10, shown as orange pentagons), the Cas9 gRNA (NTLA-2001) and ddPCR probe sites. b, Relative quantification of genome-editing efficiency measured by ddPCR in HepG2 cells transfected with individual crRNAs (UT01–UT10) or the Cas9 gRNA (NTLA-2001) (n = 2–4 independent biological replicates (separately seeded and transfected HepG2 cell cultures)). c, Relative quantification of TTR mRNA expression levels by RT–qPCR using exon 1–2 and exon 3–4 junction probes. Several Cas3 crRNAs (UT05–UT07) resulted in reduced TTR transcript levels, with magnitudes comparable to or approaching NTLA-2001 in this assay (n = 3 independent biological replicates (separately seeded and transfected HepG2 cell cultures)). Error bars indicate the mean ± s.e.m. Statistical analysis was performed using a one-way ANOVA with Dunnett’s post hoc test versus untreated HepG2 cells (****P < 0.0001; NS, not significant).
Next, we compared the genome-editing profiles of Cas3 and Cas9 in HepG2 cells using WGS with both short-read and long-read platforms. Cells were transfected with either pCas3 encoding UT05 or px458 encoding NTLA-2001. At the on-target site, short-read WGS revealed 69 split reads in Cas3-treated samples, with an average deletion size of 3,851 bp and a maximum of 43,983 bp (Extended Data Fig. 9a). Among these, ten reads extended into the downstream B4GALT6 gene. Given our prior observation that mRNA–LNP delivery of Cas3 induces more confined deletions than plasmid-based delivery (Fig. 2a versus Fig. 3e), we compared off-target deletion frequencies at B4GALT6 exon 9 using ddPCR. The pCas3 plasmid-based Cas3 delivery induced 13.71% deletion at B4GALT6, whereas mRNA-based delivered Cas3 induced only 5.02%, comparable to 4.93% observed for Cas9/NTLA-2001 delivered by px458 (Extended Data Fig. 9b).
To further evaluate genome-wide specificity, we conducted short-read WGS at an average depth of 139.4× in HepG2 cells transfected with pCas3 encoding UT05 (Extended Data Fig. 10a,b). A total of 3,035 candidate deletion regions with a split-read ratio ≥ 6.44 (corresponding to estimated editing efficiency ≥ 4%) were identified, with only 15 overlapping POT sites (Extended Data Fig. 10c). Genome-wide distribution showed a single prominent deletion peak at the TTR locus, with no other notable peaks (Extended Data Fig. 10d). Integrative Genomics Viewer (IGV) analysis of POT regions confirmed that all observed signals were localized to repetitive or low-complexity regions and were not reproducible across biological replicates (Extended Data Fig. 10e). Long-read WGS further corroborated that Cas3-mediated deletions were confined to the on-target TTR site, with no structural variations detected elsewhere (Extended Data Fig. 10f,g). In contrast, short-read WGS of Cas9-edited HepG2 cells using the NTLA-2001 gRNA identified three off-target deletions—POT93 and POT200 (both intergenic) and POT88 (located in an intron of PLCB4)—among the predicted POT sites (Extended Data Fig. 10h–j). These off-target events occurred at lower frequencies than the on-target editing and were confined to noncoding or intronic regions.
Discussion
CRISPR–Cas systems have become central tools for genome engineering. Among them, class 2 Cas9 is the most widely adopted system because of its simplicity and versatility and it has advanced to clinical applications including ATTR (Supplementary Table 1). Class 1 systems such as CRISPR–Cas3, which operate through a multicomponent Cascade complex and the Cas3 helicase–nuclease, have been less studied in mammalian contexts. Their ability to induce long-range, mostly directional deletions with high sequence specificity raises interest for settings where strong gene disruption is desired, although the variability of deletion size requires careful consideration.
In this study, we evaluated Cas3 editing of TTR in vitro and in vivo, using plasmid DNA and mRNA–LNP delivery. In mouse liver, a single administration of Cas3–LNPs reduced serum TTR levels and, in exon-humanized mice, Cas3 editing decreased serum TTR levels and reduced TTR deposition with macrophage infiltration in cardiac tissue. Although occasional deletions extended into adjacent noncoding regions, no pathological changes were observed over the observation period. The long-term relevance of low-frequency deletions in noncoding regions remains to be clarified. Deletion profiles differed between delivery formats; plasmid-based expression produced wider distributions, whereas mRNA–LNP delivery was associated with more confined deletions, consistent with the transient expression window of mRNA. Most deletions followed the expected mostly directional pattern, although rare atypical events deviating from this pattern were observed (Extended Data Fig. 9a), highlighting the need for further mechanistic analysis with long-read sequencing.
Cas9 and Cas3 both edited the TTR locus efficiently but their outcomes differed. Cas9 (NTLA-2001) produced IFMs in humanized mice, including p.D39_40TdelinsA, a variant with structural features of amyloidogenic TTR66. In clinical studies, Cas9-based in vivo editing (NTLA-2001) achieved sustained plasma TTR reduction of up to 90% over 24 months with a favorable safety profile18. Cas3 did not generate detectable IFMs under our conditions, instead producing larger and more variable deletions. The implications of these distinct editing outcomes merit further investigation. In HepG2 cells, ddPCR analysis showed that selected Cas3 crRNAs (for example, UT05) achieved editing efficiencies similar to those of the Cas9 guide NTLA-2001 (refs. 16,17) (Fig. 6). WGS analyses showed no reproducible off-target edits with Cas3 under the tested conditions, while Cas9 produced low-frequency events at three loci located in intronic or intergenic regions (Extended Data Fig. 10h–j). Beyond gene disruption, catalytically inactivated Cascade complexes have been adapted for transcriptional and epigenetic regulation in both plant and mammalian systems44,45. Together with base and prime editors, the growing repertoire of class 1 and class 2 editors illustrates the diversification of genome-editing modalities, enabling researchers to match each system to specific biological or therapeutic aims16,17,19,21,24,35,36,67,68,69,70,71,72.
Taken together, our results show that Cas3 can mediate efficient editing with a characteristic but variable long-range deletion profile in human cells and mouse models. Although Cas9 remains the benchmark for clinical genome editing, Cas3 provides a mechanistically distinct and complementary approach. Further optimization of delivery methods and comprehensive safety studies will be needed to clarify its potential and limitations.
Methods
crRNA design and plasmid construction
In the type I-E CRISPR–Cas3 system, CRISPR arrays are transcribed as a long precursor (pre-crRNA) composed of repeat–spacer–repeat units. Each repeat sequence forms a stable stem loop that is specifically recognized and cleaved by the Cas6 endoribonuclease. Cas6 processing generates individual mature crRNAs, each typically consisting of an 8-nt 5′ handle derived from the repeat, a 32-nt spacer that specifies the DNA target and a 21-nt 3′ handle derived from the repeat hairpin. After cleavage, Cas6 remains bound to the 3′ hairpin and the mature crRNA is incorporated into the Cascade surveillance complex. Within Cascade, Cas5 binds the 5′ handle, multiple Cas7 subunits align along the spacer to form the backbone, Cas6 caps the 3′ end and Cas8 and Cas11 complete the complex assembly. This arrangement is essential for target recognition and recruitment of Cas3 for unidirectional DNA degradation50.
The all-in-one plasmid (pRB-EF1a-Cas3-Cascade-U6v2-BbsI-GFP: pCas3) expresses the crRNA and Escherichia coli Cascade–Cas3 complex along with EGFP under the control of the EF1a promoter, ensuring strong and constitutive expression (Extended Data Fig. 2 and Supplementary Data 1). Each Cas protein is linked by 2A self-cleaving peptides, enabling efficient polycistronic expression, and the transcript terminates with a poly(A) signal to enhance mRNA stability and translation. All coding sequences downstream of the EF1α promoter were codon-optimized for mammalian expression. The plasmid was custom-synthesized by VectorBuilder on the basis of our design. Five crRNAs targeting the first or second exons of the mouse Ttr gene (ENSMUST00000075312.5) were designed (Supplementary Table 2). For each crRNA, two complementary 5′-phosphorylated oligonucleotides encompassing the crRNA sequence and BbsI restriction endonuclease site overhangs were synthesized, annealed, subcloned into a pCas3-Cascade-GFP plasmid digested with BbsI-HF (R3539L, New England Biolabs) and purified using NucleoSpin gel and PCR cleanup (740609, Macherey-N-nagel). G211 sgRNA for CRISPR–Cas9 was similarly subcloned into a pSpCas9(BB)-2A-GFP plasmid (pX458; Addgene, 48138). The resultant constructs were subjected to Sanger sequencing to verify proper sub-cloning of the crRNA or sgRNA sequence.
Cell culture
The mouse hepatoma cell line Hepa1-6 (BRCB1638) and the human hepatocellular carcinoma cell line HepG2 (RCB1886) were obtained from the RIKEN Cell Bank. MEFs were collected at E14.5 from the interbreeding of TtrhV30orf/hV30orf mice (Transgenic).
The cells were cultured in DMEM (08459-64, Nacalai Tesque, for Hepa1-6; 11885-084, Thermo Fisher Scientific, for HepG2) supplemented with 10% heat-inactivated FBS (A4736401, Thermo Fisher Scientific) and 0.1 mg ml−1 penicillin and streptomycin (26253-84, Nacalai Tesque) on collagen-I-coated dishes (IWAKI) at 37 °C in a humidified atmosphere with 5% CO2.
Plasmid transfection and FACS isolation of GFP-expressing cells
pCas3 or px458 plasmids were transfected by Lipofectamine 3000 (Thermo Fisher Scientific). Then, 48 h after lipofection, cells were subjected to FACS isolation of GFP+ cells using BD FACSAria Soap (BD Biosciences). Briefly, cells were detached using trypsin–EDTA solution (32777-44, Nacalai Tesque), then passed through a 35-μm cell strainer to achieve a single-cell suspension and sorted for GFP fluorescence, using nontransfected Hepa1-6 cells to define the background fluorescence level. Flow cytometry data were analyzed with FlowJo software (version 10.10.0, BD Biosciences). Gates were set on the basis of forward and side scatter to exclude debris and doublets, while GFP⁺ populations were defined using untransfected cells as negative controls. A representative gating strategy used for the isolation of GFP⁺ cells is shown in Supplementary Fig. 3. After FACS isolation, cells were harvested. gDNA was extracted using NucleoSpin Tissue XS (740901, Macherey-Nagel) and RNA was isolated using an RNeasy mini kit (74104, Qiagen).
Evaluation of genome-editing efficiency
PCR primers were designed around the target site (Supplementary Table 6) and the region was amplified using Quick Taq HS DyeMix (DTM-101, Toyobo), followed by electrophoresis on agarose gel.
To compare the genome-editing efficiency in a quantitative manner, ddPCR was performed. Primer and probe sequences are listed in Supplementary Table 7. TaqMan copy number reference assay, mouse, Tfrc and human RNaseP (Thermo Fisher Scientific) were used as reference. After PCR amplification, plates were transferred into the Bio-Rad QX-200 droplet reader. All assays were analyzed using the QX-200 droplet reader and Quantasoft Analysis Pro (Bio-Rad).
For RT–qPCR, RNA was subjected to RT using ReverTra Ace qPCR RT master mix with gDNA remover (FSQ-301, Toyobo). qPCR was performed using SsoAdvanced Universal SYBR green supermix (Bio-Rad) on an CFX96 deep-well real-time PCR detection system (Bio-Rad). Primers used for RT–qPCR are listed in Supplementary Table 8. The mean cycle threshold (Ct) of target gene (Ttr) expression was normalized to that for the control gene, mouse B2m or human GAPDH.
WGS with short-read and long-read sequencing
gDNA was extracted from pCas3-cr2-transfected Hepa1-6 and HepG2 cells. The TruSeq DNA PCR-free library kit (Illumina) was used to prepare the library for short-read sequencing. Next-generation sequencing (NGS) was conducted on a NovaSeq X Plus platform (2 × 150 bp) at Macrogen. Raw sequencing reads were processed following standard bioinformatics pipelines. Reads were mapped to the reference mouse genome (GRCm39) or human genome (GRCh38) and large deletions were identified using split-read and discordant-read analyses. Candidate off-target sites were filtered on the basis of split-read counts exceeding five and base-coverage ratios above 0.2. The relative split-read count ratio between Cas3-treated and control samples was calculated to identify high-confidence off-target loci, which were visualized using IGV software.
For long-read sequencing, sequencing libraries were prepared using the ligation sequencing kit V14 (Oxford Nanopore Technologies) and sequenced on a PromethION P2 solo sequencer equipped with a FLO-PRO114M flow cell and the SQK-LSK114 library preparation kit. Basecalling was performed with Dorado version 7.3 (super accuracy model version 5.0.0). Quality trimming was conducted using BBDuk (BBtools version 39.13) to remove bases with Q scores < 15 at read ends. Reads with Q scores ≥ 15 and lengths ≥ 100 bp were retained. Sequencing statistics are as follows. Mouse (Hepa1-6): Cas3, 10.1 million reads, 90.7 Gb, median Q score = 25.5; Cas9, 4.8 million reads, 33.8 Gb, Q score = 26.0; control, 4.0 million reads, 27.5 Gb, Q score = 27.1. Human (HepG2): UT05, 6.1 million reads, 86.7 Gb, Q score = 28.1; NTLA (Cas9), 7.7 million reads, 98.9 Gb, Q score = 28.1; control, 4.7 millionM reads, 73.0 Gb, Q score = 27.7. Reads were mapped using Minimap2 version 2.1 to GRCm39 (mouse) or GRCh38 (human). Cas3-mediated deletions (100 bp–1 Mb) were detected using Sniffles version 2.6.2 with the ‘--no-qc’ option. Germline deletions, defined as recurrent identical deletions (n ≥ 3) across all samples, were excluded along with flanking regions ±1 kb. To minimize false positives, we excluded (1) reads with MAPQ < 60; (2) reads with >5 deletions; (3) regions with ≥50 consecutive Ns and surroundings ±10 kb; and (4) regions with >3× median coverage, coverage < 10 or located within ±100 bp of deletions showing a secondary alignment rate ≥ 0.05 or mismatch rate ≥ 0.01. A 100-kb Cas3 target region (10 kb upstream, 90 kb downstream of the recognition site) was defined. Deletions within this region were evaluated after coverage normalization.
CapSeq of off-target analysis at POT of Cas3 and Cas9
Targeted regions were selected as follows. Probes around on-target regions at Ttr covered 90 kb upstream to 10 kb downstream of the PAM. Probes around POT regions of CRISPR–Cas3 covered 4.5 kb upstream to 0.5 kb downstream of the potential PAM. Probes of CRISPR–Cas9 covered 1 kb upstream and downstream of the potential PAM. For NGS, gDNA was extracted from transfected Hepa1-6 cells and sheared with SureSelect enzymatic fragmentation kit (Agilent). After preparation of the DNA library with SureSelectXT-HS2 reagents (Agilent) and custom probe kit designed by SureDesign, genomic sequence analysis was performed using HiSeq X (2 × 150 bp) according to the standard procedure at Macrogen. Discordant reads and split reads were extracted by SAMtools and Lumpy-sv, respectively, and the total number of each read at on-target or POT regions was counted using BEDTools. Mutation patterns at the on-target locus were analyzed from extracted discordant reads mapped at deletion hotspots predicted by split-read counts of every 100 bp of on-target locus. For off-target effects at POT regions, split-read counts were normalized by total read counts of each region and calculated by subtracting read counts of control samples from those of CRISPR-transfected samples. Small indel mutations around PAM sequences, which were undetected using the aforementioned methods, were analyzed by CRISPResso2 (http://crispresso.pinellolab.partners.org/) according to the developers’ protocol73.
RNA transfection
GFP, Cascade (Cas5, Cas6, Cas7, Cas8 and Cas11)–Cas3 and Cas9 mRNAs with modifications including Cap1 and/or N1mΨ triphosphate, 5Me-cytidine triphosphate and 5Mo-uridine triphosphate were synthesized by Elixirgen Scientific. crRNAs containing 2′-O-methyl and phosphorothioate modifications were synthesized by Integrated DNA Technologies. RNAs were introduced into approximately 80% confluent Hepa1-6, HepG2 and MEF cells in six-well plates using Lipofectamine MessengerMAX (LMRNA001, Thermo Fisher Scientific) according to the manufacturer’s instructions. An Attune NxT flow cytometer (Invitrogen) was used to detect GFP signals. Viability of transfected cells was evaluated by propidium iodide staining.
LNP formulation
mRNA was loaded into LNPs using microfluidic mixing methods74. An ethanol solution containing an ionizable lipid, 1,2-distearoyl-sn-glycero-3-phosphocholine, cholesterol and PEG-DMG at fixed molar ratios (50:3:47:1.5) was prepared at a total lipid concentration of 8 mM. Cas9 was loaded into LNPs at a weight ratio of 1:1 for Cas9 mRNA:sgRNA. For Sep-Cas3, Cas3, Cas5, Cas6, Cas7, Cas8, Cas11 and crRNA were incorporated at a weight ratio of 1:1:1:1:1:1:1. For DiT-Cas3, Cas3–Cas5–Cas11, Cas6–Cas7–Cas8 and crRNA were loaded at a weight ratio of 3:3:1, respectively. The RNA cargo was dissolved in 50 mM citrate buffer (pH 4.0), resulting in a final RNA cargo concentration of 81.5 µg ml−1. The lipid ethanol solution and RNA solution were rapidly mixed using a glass-based iLiNP device75 at a total flow rate of 5 ml min−1 and RNA-to-lipid flow rate of 3. The nitrogen-to-phosphate ratio was adjusted to 6. The resulting LNP solution was dialyzed for 2 h or more at 4 °C against 20 mM Tris-HCl buffer (9% sucrose, pH 7.40) using Slide-A-Lyzer G3 dialysis cassettes (molecular weight cutoff (MWCO): 20 kDa; Thermo Fisher Scientific). The LNP solution was concentrated by ultrafiltration using an Amicon Ultra-15 unit (MWCO: 100 kDa; Millipore). The size and polydispersity of LNPs were measured by a Zetasizer Nano ZS ZEN3600 instrument (Malvern Instruments). The encapsulation efficiency and total concentration of mRNA were measured by a Ribogreen assay76.
LNP delivery in vivo
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Tokyo (approval no. A21-57). Male Jcl:ICR (CLEA), TtrhV30orf/hV30orf (Transgenic) mice were kept under specific-pathogen-free conditions until use. Only male mice were used because serum TTR concentrations were consistently lower in females, which could introduce variability in data interpretation. Using male mice ensured consistency across experimental groups. All mice were housed in standard cages under a 12-h light–dark cycle (lights on from 7:00 a.m. to 7:00 p.m.) at a constant temperature of 22 ± 2 °C and relative humidity of 40–60%. Animals were provided with standard rodent chow and water ad libitum.
LNPs and saline as negative control were dosed through the lateral tail vein. Blood was collected into serum separator tubes (MiniCollect II, Greiner Bio-One) for circulating TTR quantitation and cytokine measurements. Tissue from the liver, spleen and kidney was collected for DNA and/or RNA extraction. Primary hepatocytes were isolated with the gentleMACS perfusion technology (Miltenyi Biotec) according to the manufacturer’s protocol.
TTR ELISA analysis
Total TTR serum levels were determined using a mouse prealbumin (TTR) ELISA kit (Aviva Systems Biology, OKIA00111) for Jcl:ICR mice, and human prealbumin (TTR) AssayMax ELISA kit (AssayPro, EP3010-1) for TtrhV30orf/hV30orf mice. Serum was diluted 1,000–10,000-fold for mouse Ttr and 4,000–8,000-fold for human TTR. Plates were read on a SYNERGY LX multimode reader (Bio Tek) at an absorbance of 450 nm. Serum TTR levels were calculated by Gen5 software (version 3.09). Final serum values were adjusted for assay dilution.
Histological analysis
Mice were deeply anesthetized by isoflurane and then perfused transcardially with 4% paraformaldehyde (PFA). Liver samples were postfixed with 4% PFA. Paraffin-embedded liver sections (4 μm thick) were stained with DFS and anti-mouse Ttr antibody (1:250; LS-C407961, LSBio). Frozen liver sections (4 μm thick) were stained with anti-TTR antibody (1:1,000; 201630-T10, SinoBiological), and anti-CD68 antibody (1:50; ab53444, Abcam)49,63. Images were acquired using a BZ-X700 fluorescence microscope (Keyence) and BZ-X-Analyzer software (Keyence).
Cas3 western blotting
Liver tissue samples of mice were flash-frozen in liquid nitrogen and homogenized in 2× cell lysis buffer (Cell Signaling) supplemented with cOmplete protease inhibitor (Sigma-Aldrich) using a TissueLyser II (Qiagen) according to the manufacturer’s instructions. Homogenized samples were incubated on ice for 30 min and centrifuged at 100g for 30 min at 4 °C. Supernatants were used as lysate samples. Total protein samples (30 μg) were separated on 4–12% Bolt Bis–Tris plus mini protein gels (Invitrogen) and transferred to polyvinylidene difluoride membranes (Invitrogen). Cas3 was detected with rat polyclonal anti-Cas3 (1:1,000, C4U Corporation), followed by anti-rat IgG, horseradish peroxidase (HRP)-linked antibody (1:2,000; 7077, Cell Signaling). α-Tubulin was detected with α-tubulin Antibody (1:2,000; Cell Signaling, 7077 and 2144), followed by anti-rabbit IgG, HRP-linked antibody (1:2,000; Cell Signaling, 7074). Signals were detected using SuperSignal WestPico plus chemiluminescence substrate (Thermo Fisher Scientific) and densitometry analysis was performed using iBright CL 1500 (Thermo Fisher Scientific).
Cytokine analysis
First, 50–100 μl of blood was collected by tail-vein nick for serum cytokine measurements at 4, 24 and 72 h and 1 week after administration. The enhanced chemiluminescence multiplex assay (U-PLEX custom biomarker group 1 (mouse) assays, K15069M-1) was used for cytokine analysis at the Research Center for Immunological Analysis. Serum was diluted threefold using sample diluent 41. Data were analyzed using Discovery Workbench 4.0 (Meso Scale Discovery).
Amplicon sequencing
The ratio of IFMs generated by Cas9 was analyzed by NGS of PCR amplicons. gDNA was extracted from MEFs isolated from TtrhV30orf/hV30orf mice and the 240-bp region including estimated cut site was amplified by PCR. Primer sequences are listed in Supplementary Table 9. Amplicons from PCR were purified by column purification with NucleoSpin gel and PCR cleanup kit (Macherey-Nagel) and sequenced by MiSeq (2 × 250 bp) according to a standard procedure at the NGS Core facility at Osaka University. Raw reads from each sample were analyzed by the ratio of modified to unmodified reads using CRISPResso2 (http://crispresso.pinellolab.partners.org/) according to the developers’ protocol73.
nanoLC–MS/MS analysis
Each serum sample was lysed in 8 M urea, reduced with 1 mM dithiothreitol for 90 min and alkylated with 5.5 mM iodoacetamide for 30 min. After digestion with MS-grade lysyl endopeptidase (Fuji Film Wako Chemicals) at 37 °C for 3 h, the resulting peptide mixtures were diluted with 10 mM Tris-HCl pH 8.2 to achieve a final concentration of <2 M urea and subsequently digested with MS-grade trypsin gold (Promega) at 37 °C for 3 h. An equal amount of trypsin was added for overnight digestion and fragmented peptides were desalted using ZipTip C18 (Millipore). Shotgun proteomic analyses were performed by Orbitrap Eclipse Tribrid MS instrument with FAIMS Pro interface (Thermo Fisher Scientific) connected to a Vanquish Neo ultrahigh-performance LC system (Thermo Fisher Scientific). Peptide samples were separated using a linear gradient of 2–24% mobile phase (0.1% formic acid in acetonitrile) at 300 nl min−1. Full-scan MS spectra were acquired at a resolution of 120,000 and subsequent MS/MS scans were performed in the ion trap using collision-induced dissociation fragmentation with a normalized collision energy of 35% with 10 ms of maximum injection time. Protein identification was conducted by searching against the customized database of UniProt mouse reference proteome (UP000000589) and WT/IFM-derived TTR amino acid sequence data (Fig. 5b and Extended Data Figs. 6 and 7) using the Sequest HT algorithm in Proteome Discoverer Software (version 2.5; Thermo Fisher Scientific).
Recombinant TTR expression and purification
WT and two mutant forms of TTR (V30M and IFM4) were expressed in an E. coli expression system. The overexpressed proteins were purified using a nickel affinity resin (Qiagen), followed by size-exclusion chromatography with a HiLoad 10/300 Superdex 75-pg column (Cytiva) running on an ÄKTA pure 25 system (Cytiva).
Aggregation assay
Under two pH conditions (pH 7.5 and 4.5) at 37 °C, TTR precipitated as large aggregates that can be measured by turbidity using the Prometheus Panta (NanoTemper Technologies GmbH). The turbidity of the protein solution was recorded over a period of 72 h to monitor aggregation kinetics. For TTR tetramer stabilization studies, TTR aggregates were also monitored by SDS–PAGE. Specifically, TTR samples (0.5 mg ml−1) were incubated in acetate buffer (100 mM Tris, 50 mM acetate and 100 mM KCl, pH 4.5–7.0) for 72 h at 37 °C. The TTRs were immediately processed under nonreducing conditions and were loaded on 4–12% Bis–Tris plus mini protein gels (Invitrogen). Protein bands were visualized by Coomassie brilliant blue staining (Nacalai Tesque).
Statistics and reproducibility
Statistical analysis was performed using GraphPad Prism 10 software. All results are presented as the mean ± s.e.m. Comparisons between two groups were performed using an unpaired two-tailed Student’s t-test for parametric data or two-tailed Student’s t-test with Welch’s correction if the s.d. was not equal. Comparisons among multiple groups were analyzed by one-way analysis of variance (ANOVA). All experiments were independently repeated at least three times with similar results. Representative images shown in Fig. 4b and Extended Data Figs. 4c, 6b,i and 8a,e,f are from experiments that yielded comparable outcomes. All relevant experimental conditions, including age, sex, body weight and culture conditions, were controlled and standardized across all experimental groups. Therefore, consideration of additional covariates was not applicable for this study.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. WGS, capture and amplicon sequencing data generated in this study were deposited to the DNA Data Bank of Japan Sequence Read Archive under BioProject accession PRJDB37936 and are publicly accessible. All other data supporting the findings of this study are available within the paper and its Supplementary Information. Source data are provided with this paper.
References
Koike, H. & Katsuno, M. Transthyretin amyloidosis: update on the clinical spectrum, pathogenesis, and disease-modifying therapies. Neurol. Ther. 9, 317–333 (2020).
Porcari, A., Fontana, M. & Gillmore, J. D. Transthyretin cardiac amyloidosis. Cardiovasc. Res. 118, 3517–3535 (2022).
Rapezzi, C. et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat. Rev. Cardiol. 7, 398–408 (2010).
Adams, D., Koike, H., Slama, M. & Coelho, T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat. Rev. Neurol. 15, 387–404 (2019).
Sekijima, Y. et al. The biological and chemical basis for tissue-selective amyloid disease. Cell 121, 73–85 (2005).
Sikora, J. L. et al. Genetic variation of the transthyretin gene in wild-type transthyretin amyloidosis (ATTRwt). Hum. Genet. 134, 111–121 (2015).
Cornwell, G. G., Murdoch, W. L., Kyle, R. A., Westermark, P. & Pitkänen, P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am. J. Med 75, 618–623 (1983).
Tanskanen, M. et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in α2-macroglobulin and tau: a population-based autopsy study. Ann. Med. 40, 232–239 (2008).
Kotit, S. Lessons from the first-in-human in vivo CRISPR/Cas9 editing of the TTR gene by NTLA-2001 trial in patients with transthyretin amyloidosis with cardiomyopathy. Glob. Cardiol. Sci. Pr. 2023, e202304 (2023).
Aimo, A. et al. RNA-targeting and gene editing therapies for transthyretin amyloidosis. Nat. Rev. Cardiol. 19, 655–667 (2022).
Tschöpe, C. & Elsanhoury, A. Treatment of transthyretin amyloid cardiomyopathy: the current options, the future, and the challenges. J. Clin. Med. 11, 2148 (2022).
Adams, D. et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 379, 11–21 (2018).
Adams, D. et al. Hereditary transthyretin amyloid neuropathies: advances in pathophysiology, biomarkers, and treatment. Lancet Neurol. 22, 1061–1074 (2023).
Chavez, M., Chen, X., Finn, P. B. & Qi, L. S. Advances in CRISPR therapeutics. Nat. Rev. Nephrol. 19, 9–22 (2023).
Doudna, J. A. The promise and challenge of therapeutic genome editing. Nature 578, 229–236 (2020).
Gillmore, J. D. et al. CRISPR–Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 385, 493–502 (2021).
Fontana, M. et al. CRISPR–Cas9 gene editing with nexiguran ziclumeran for ATTR cardiomyopathy. N. Engl. J. Med. 391, 2231–2241 (2024).
Gillmore, J. D. et al. Nexiguran ziclumeran gene editing in hereditary ATTR with polyneuropathy. N. Engl. J. Med. 393, 1375–1386 (2025).
Longhurst, H. J. et al. CRISPR–Cas9 in vivo gene editing of KLKB1 for hereditary angioedema. N. Engl. J. Med. 390, 432–441 (2024).
Intellia Therapeutics. NTLA-3001 in Adults with Alpha-1 Antitrypsin Deficiency-Associated Lung Disease. https://clinicaltrials.gov/study/NCT06622668 (2025).
Pierce, E. A. et al. Gene Editing for CEP290-Associated Retinal Degeneration. N. Engl. J. Med. 390, 1972–1984 (2024).
Excision BioTherapeutics. Study of EBT-101 in Aviremic HIV-1 Infected Adults on Stable ART. https://clinicaltrials.gov/study/NCT05144386 (2024).
CRISPR Therapeutics. CRISPR Therapeutics Reports Positive Additional Phase 1 Data for CTX310 Targeting ANGPTL3 and Provides Update on In Vivo Cardiovascular Pipeline. Press Release. Available at https://crisprtx.com/about-us/press-releases-and-presentations/crispr-therapeutics-reports-positive-additional-phase-1-data-for-ctx310-targeting-angptl3-and-provides-update-on-in-vivo-cardiovascular-pipeline (2025).
Morrow, P. K. et al. CTX320: an investigational in vivo CRISPR-based therapy efficiently and durably reduces lipoprotein (a) levels in non-human primates after a single dose. Circulation 148, A17013 (2023).
Regeneron Pharmaceuticals. A Study to Investigate the Safety and Effectiveness of a Coagulation Factor IX Gene Insertion Therapy (REGV131-LNP1265) in Pediatric, Adolescent and Adult Participants With Hemophilia B. https://clinicaltrials.gov/study/NCT06379789 (2025).
Verve Therapeutics, Inc. A Study of VERVE-101 in Patients With Familial Hypercholesterolemia and Cardiovascular Disease. https://clinicaltrials.gov/study/NCT05398029 (2025).
Verve Therapeutics, Inc. A Study of VERVE-102 in Patients with Familial Hypercholesterolemia or Premature Coronary Artery Disease. https://clinicaltrials.gov/study/NCT06164730 (2024).
Verve Therapeutics, Inc. Phase 1b Study of VERVE-201 in Patients With Refractory Hyperlipidemia. https://clinicaltrials.gov/study/NCT06451770 (2025).
Beam Therapeutics Inc. A Study to Evaluate the Safety and Efficacy of BEAM-302 in Adult Patients With Alpha-1 Antitrypsin Deficiency (AATD). https://clinicaltrials.gov/study/NCT06389877 (2025).
Arbor Biotechnologies. Phase 1/2 Study of ABO-101 in Primary Hyperoxaluria Type 1 (redePHine). https://clinicaltrials.gov/study/NCT06839235 (2025).
iECURE, Inc. An Open-Label Study to Investigate ECUR-506 in Male Babies Less Than 9 Months of Age With Neonatal Onset OTC Deficiency. https://clinicaltrials.gov/study/NCT06255782 (2025).
Precision BioSciences, Inc. Phase 1 Study to Evaluate Safety and Antiviral Activity of PBGENE-HBV in Adult Patients with Chronic Hepatitis B. https://clinicaltrials.gov/study/NCT06680232 (2025).
Tune Therapeutics, Inc. Phase 1b, Open-Label Study of Tune-401 to Assess Safety, PK and PD in Adults With Chronic Hepatitis B. https://clinicaltrials.gov/study/NCT06671093 (2025).
Sangamo Therapeutics. A Multicenter Phase 1 Double-Blind, Randomized, Sham-Controlled Dose Escalation Study to Determine Safety and Tolerability of Single Dose Intrathecal ST-503 Gene Therapy for Refractory Pain Due to Idiopathic Small Fiber Neuropathy (iSFN). https://clinicaltrials.gov/study/NCT06980948 (2025).
Kim, P. et al. Safety, pharmacokinetics, and pharmacodynamics of LBP-EC01, a CRISPR–Cas3-enhanced bacteriophage cocktail, in uncomplicated urinary tract infections due to Escherichia coli (ELIMINATE): the randomised, open-label, first part of a two-part phase 2 trial. Lancet Infect. Dis. 24, 1319–1332 (2024).
Musunuru, K. et al. Patient-specific in vivo gene editing to treat a rare genetic disease. N. Engl. J. Med. 392, 2235–2243 (2025).
Guo, C., Ma, X., Gao, F. & Guo, Y. Off-target effects in CRISPR/Cas9 gene editing. Front. Bioeng. Biotechnol. 11, 1143157 (2023).
Kosicki, M., Tomberg, K. & Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36, 765–771 (2018).
Morisaka, H. et al. CRISPR–Cas3 induces broad and unidirectional genome editing in human cells. Nat. Commun. 10, 5302 (2019).
Cameron, P. et al. Harnessing type I CRISPR–Cas systems for genome engineering in human cells. Nat. Biotechnol. 37, 1471–1477 (2019).
Csörgő, B. et al. A compact Cascade–Cas3 system for targeted genome engineering. Nat. Methods 17, 1183–1190 (2020).
Lu, M. et al. Structure and genome editing of type I-B CRISPR–Cas. Nat. Commun. 15, 4126 (2024).
Dolan, A. E. et al. Introducing a spectrum of long-range genomic deletions in human embryonic stem cells using type I CRISPR–Cas. Mol. Cell 74, 936–950.e5 (2019).
Young, J. K. et al. The repurposing of type I-E CRISPR–Cascade for gene activation in plants. Commun. Biol. 2, 1–7 (2019).
Guo, J. et al. Engineered minimal type I CRISPR–Cas system for transcriptional activation and base editing in human cells. Nat. Commun. 15, 7277 (2024).
Pickar-Oliver, A. et al. Targeted transcriptional modulation with type I CRISPR–Cas systems in human cells. Nat. Biotechnol. 37, 1493–1501 (2019).
Yoshimi, K. et al. Genome editing using type I-E CRISPR–Cas3 in mice and rat zygotes. Cell Rep. Methods 4, 100833 (2024).
Ran, F. A. et al. Genome engineering using the CRISPR–Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Finn, J. D. et al. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 22, 2227–2235 (2018).
Yoshimi, K. et al. Dynamic mechanisms of CRISPR interference by Escherichia coli CRISPR–Cas3. Nat. Commun. 13, 4917 (2022).
Bae, S., Park, J. & Kim, J.-S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014).
Pardi, N. et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 217, 345–351 (2015).
Galloway, A. & Cowling, V. H. mRNA cap regulation in mammalian cell function and fate. Biochim. Biophys. Acta 1862, 270–279 (2019).
Khvorova, A. & Watts, J. K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 35, 238–248 (2017).
Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 24, 374–387 (2014).
Inagaki, M. et al. Cap analogs with a hydrophobic photocleavable tag enable facile purification of fully capped mRNA with various cap structures. Nat. Commun. 14, 2657 (2023).
Li, Z. et al. TTR exon-humanized mouse optimal for verifying new therapies for FAP. Biochem. Biophys. Res. Commun. 599, 69–74 (2022).
Kozuka-Hata, H. et al. Real-time search-assisted multiplexed quantitative proteomics reveals system-wide translational regulation of non-canonical short open reading frames. Biomolecules 13, 979 (2023).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Dasari, A. K. R., Hung, I., Gan, Z. & Lim, K. H. Two distinct aggregation pathways in transthyretin misfolding and amyloid formation. Biochim Biophys. Acta 1867, 344–349 (2019).
Ibrahim, R. B., Liu, Y.-T., Yeh, S.-Y. & Tsai, J.-W. Contributions of animal models to the mechanisms and therapies of transthyretin amyloidosis. Front. Physiol. 10, 338 (2019).
Teixeira, C., Martins, H. S. & Saraiva, M. J. Cellular environment of TTR deposits in an animal model of ATTR-cardiomyopathy. Front. Mol. Biosci. 10, 1144049 (2023).
Buxbaum, J. N. & Reixach, N. Transthyretin: the servant of many masters. Cell. Mol. Life Sci. 66, 3095–3101 (2009).
Suenaga, G. et al. Involvement of macrophages in the pathogenesis of familial amyloid polyneuropathy and efficacy of human iPS cell-derived macrophages in its treatment. PLoS ONE 11, e0163944 (2016).
Schmidt, M. et al. Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis. Nat. Commun. 10, 5008 (2019).
Qiu, M. et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proc. Natl Acad. Sci. USA 118, e2020401118 (2021).
Lee, J. H. et al. In vivo genome editing for hemophilia B therapy by the combination of rebalancing and therapeutic gene knockin using a viral and non-viral vector. Mol. Ther. Nucleic Acids 32, 161–172 (2023).
Lee, R. G. et al. Efficacy and safety of an investigational single-course CRISPR base-editing therapy targeting PCSK9 in nonhuman primate and mouse models. Circulation 147, 242–253 (2023).
Kasiewicz, L. N. et al. GalNAc-lipid nanoparticles enable non-LDLR dependent hepatic delivery of a CRISPR base editing therapy. Nat. Commun. 14, 2776 (2023).
Packer, M. S. et al. Evaluation of cytosine base editing and adenine base editing as a potential treatment for alpha-1 antitrypsin deficiency. Mol. Ther. 30, 1396–1406 (2022).
Lek, A. et al. Death after high-dose rAAV9 gene therapy in a patient with Duchenne’s muscular dystrophy. N. Engl. J. Med. 389, 1203–1210 (2023).
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
Okuda, K. et al. On the size-regulation of RNA-loaded lipid nanoparticles synthesized by microfluidic device. J. Control. Release 348, 648–659 (2022).
Maeki, M. et al. Mass production system for RNA-loaded lipid nanoparticles using piling up microfluidic devices. Appl. Mater. Today 31, 101754 (2023).
Hashiba, K. et al. Overcoming thermostability challenges in mRNA–lipid nanoparticle systems with piperidine-based ionizable lipids. Commun. Biol. 7, 1–13 (2024).
Acknowledgements
This research was supported in part by the Japan Agency for Medical Research and Development (grant no. JP23bm1223009h0001) to T.M. and Japan Society for the Promotion of Science KAKENHI (grant no. 23H00367) to T.M. We thank J. Imamura for expert technical support related to mouse maintenance and T. Denda for help with the histological analysis. We also thank Shin-Etsu Chemical for kindly providing glass-based iLiNP microfluidic devices.
Author information
Authors and Affiliations
Contributions
S.I. and T.M. designed the experiments. S.I., K.C. and E.E. conducted the molecular biological experiments. S.I. and Y.Y. conducted the animal experiments. Y. Sato, R.I., R.S. and M.M. generated the LNPs. M.O. and H.K.-H. performed the nanoLC–MS/MS analysis. T.I. and K. Yamamura generated the TTR exon-humanized mice. K.T. characterized the mutant TTR protein. K.C., K. Yamaguchi and Y.K. performed the sequencing analysis. Y. Sekijima performed the histological analysis. F.H., Y.L., N.A. and H.A. generated the PureCap Cap2 mRNA. K. Yoshimi designed the Cascade–Cas3 mRNA.
Corresponding author
Ethics declarations
Competing interests
T.M. and K. Yoshimi are the cofounder and scientific advisor of C4U Corporation, respectively. K.C. is an employee of C4U Corporation. K. Yamamura is a scientific adviser of Transgenic Group Inc. T.I. is a director of Transgenic Inc. The other authors declare no competing interests.
Peer review
Peer review information
Nature Biotechnology thanks Chase Beisel and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 All-in-one plasmid for crRNA screening.
Schematic representation of the plasmid pRB-EF1α-Cas3Cascade-U6v2-BbsI-GFP (pCas3), used for evaluating crRNA activity in mouse Hepa1-6 cells. This all-in-one construct contains the full Cascade-Cas3 under the control of the dual EF1α promoters, followed by a codon-optimized SV40 nuclear localization signal (SV40_NLS) and GFP reporter. The crRNA expression cassette is driven by the U6 promoter with a BbsI cloning site for spacer insertion. All coding sequences downstream of the EF1α promoter were codon-optimized for efficient expression in mammalian systems. The full annotated plasmid sequence is available in Supplementary Data 1.
Extended Data Fig. 2 Evaluation of off-target effects of CRISPR–Cas3 using capture sequencing.
(a) Distribution of deletion start sites in Hepa1-6 cells transfected with the Cas3-Cascade expression plasmid (pCas3-crRNA#2), as determined by capture sequencing. (b) Similar analysis of deletion start sites in the liver of ICR mice injected with DiT-Cas3-LNP (6 mg/kg). In both panels, the horizontal axis represents deletion size (in base pairs), and the vertical axis indicates the genomic position of the deletion start site, measured relative to the crRNA binding site (position = 0). (c) Analysis of split reads at potential off-target (POT) sites in Cas3-treated cells. To evaluate off-target activity of Cas3, capture sequencing was performed for 5 kb regions flanking in silico–predicted POT sites (see Fig. 2a and Supplementary Table 7). Split reads detected at POT93 and POT295 were aligned to the mouse genome (mm10) and visualized using Integrative Genomics Viewer (IGV). RepeatMasker tracks revealed that these regions are located near repetitive elements, which may cause misalignment artifacts. Notably, the crRNA spacer sequence showed low homology to these regions, and deletion patterns were not reproducible across replicates. These observations suggest that the weak signals at POT93 and POT295 are likely false positives, rather than genuine Cas3-induced events.
Extended Data Fig. 3 Evaluation of genome-wide off-target effects of CRISPR–Cas3 using short- and long-read whole-genome sequencing.
Short-read WGS (a–d). (a) Summary of whole-genome sequencing (WGS) coverage and mapping statistics for pCas3-crRNA#2–transfected Hepa1-6 cells and untransfected controls. Sequencing depth averaged 139.5×, providing high sensitivity for detecting structural variants. (b) Number of candidate deletion regions associated with Cas3 activity. A total of 1,037 genomic regions were identified with a split-read ratio ≥ 1.38, corresponding to an estimated editing efficiency ≥ 10%. Among these, 12 regions overlapped with potential off-target (POT) sites previously evaluated by capture sequencing. (c) IGV snapshot showing representative aligned reads with deletions at the intended Ttr locus, confirming on-target activity of Cas3. (d) IGV snapshot of one representative candidate region among the 12 overlapping sites. All deletion signals were localized within <1 kb and mapped to low-complexity or repetitive regions, with no reproducible structural variants across replicates. These findings suggest that apparent off-target signals likely reflect alignment artifacts. Long-read WGS (e–h). (e) Quantification of candidate deletion regions identified by Oxford Nanopore long-read WGS in pCas3-crRNA#2–transfected Hepa1-6 cells and untransfected controls. (f) After excluding low-confidence or artifact-prone loci, only one candidate deletion region remained in Cas3-treated samples, corresponding to the targeted Ttr locus. (g) IGV snapshot of aligned long reads at the Ttr locus shows characteristic Cas3-induced deletions. (h) IGV snapshot of a representative excluded region illustrates that apparent deletion signals were non-reproducible, derived from poorly mappable sequences, and not supported by split reads consistent with Cas3 activity. Together, these results indicate that Cas3-mediated deletions were restricted to the intended genomic locus, with no detectable large off-target deletions elsewhere in the genome.
Extended Data Fig. 4 Evaluation of nucleotide modifications in mRNA and optimization of Cas3–Cascade RNA delivery formats.
(a) Cell viability and (b) GFP fluorescence intensity in Hepa1-6 cells transfected with in vitro transcribed GFP mRNAs incorporating different nucleotide modifications and 5′ cap structures. Tested modifications included 5-methylcytidine triphosphate (5mCTP), pseudouridine (Ψ), N1-methyl-pseudouridine (N1mΨ), and 5-methoxyuridine triphosphate (5moUTP), applied either individually or in combination. Each transcript was synthesized with either a Cap0 or Cap1 5′ cap structure. Cell viability (a) remained unaffected across all conditions, while GFP expression (b) was consistently enhanced by the presence of the Cap1 structure regardless of the internal modification pattern. Optimization of mRNA and crRNA delivery formats (c–g). (c) Agarose gel electrophoresis of PCR amplicons from Hepa1-6 cells transfected with Cas3 mRNA and crRNA in varying ratios. Distinct smaller bands indicate cleavage at the target site. Comparable editing efficiencies were observed under conditions with increased crRNA or higher total mRNA input, suggesting that Cas3 activity tolerates modest stoichiometric variation. (d) Schematic of three mRNA delivery formats: all-in-one (AiO), separated (Sep), and divide-in-two (DiT). Constructs include N- and C-terminal nuclear localization signals (NLS) and P2A self-cleaving peptides to link Cas subunits where applicable. (e) ddPCR-based quantification of genome editing efficiency in Hepa1-6 cells using AiO, Sep, or DiT mRNA configurations. DiT yielded the highest efficiency (n = 3 independent biological replicates (separately seeded and transfected Hepa1-6 cell cultures)). (f) Structure of crRNA containing base modifications at the spacer region. (g) Comparison of editing efficiencies using five chemically modified crRNAs in the DiT/Cas3 format. Pseudouridine-modified crRNA showed the highest efficiency (n = 3 independent biological replicates (separately seeded and transfected Hepa1-6 cell cultures)). Error bars represent mean ± SEM. Statistical analysis: one-way ANOVA with Dunnett’s post hoc test (vs Cas3/TTR#2).
Extended Data Fig. 5 Evaluation of Cas3–LNP efficacy, tolerability, and biodistribution in mice.
(a) Serum TTR levels at 1 week after the administration of Cascade-Cas3 mRNA-LNPs. Scores were normalized by serum TTR levels in saline injected mice. Error bars indicate the mean ± SEM. One-way ANOVA with Dunnett’s post-test (vs Saline). (n = 4-9 biological replicates per group). (b)Dose-dependent Cas3-mediated editing at the mRNA level in ICR mouse liver. Relative expression levels of Ttr mRNA in the livers of ICR mice were assessed by RT-qPCR one week after treatment with different doses of Cas3-LNPs. Error bars represent the mean ± SEM. Statistical analysis was performed using one-way ANOVA followed by Dunnett’s post hoc test (vs. saline). (n = 3 biological replicates per group). Tolerability and biodistribution of Cas3-LNP components (c-e). (c) ddPCR analysis of genome editing efficiency in spleen and kidney tissues following treatment with saline, Sep-Cas3-LNP (6 mg/kg), or DiT-Cas3-LNP (6 mg/kg). (n = 3 biological replicates per group) Statistical analysis was performed using one-way ANOVA. No significant editing was detected outside the liver. (d) Evaluation of genome editing activity in mouse liver at 4, 6, and 24 h, and 1 week after administration of saline or DiT-Cas3-LNP (6 mg/kg). PCR amplification of the Ttr target region followed by agarose gel electrophoresis revealed editing events over time. The appearance of smaller amplicons indicated the onset of genome editing within 4 h post-injection. (e) Residual Cas3 mRNA levels in liver tissue at the indicated time points after DiT-Cas3-LNP (6 mg/kg) administration, as assessed by ΔCt values relative to the reference gene B2m. Lower ΔCt values indicate higher mRNA abundance. Error bars represent the mean ± SEM. (n = 3 biological replicates per group).
Extended Data Fig. 6 Detection and characterization of a mutant TTR peptide derived from in-frame mutation (IFM) in exon-humanized mice treated with Cas9-LNP.
(a) Serum TTR levels in 1-month-old exon-humanized TTR mice measured 1 week after intravenous administration of DiT-Cas3-LNP (6 mg/kg) or Cas9/NTLA2001-LNP (2 mg/kg). Statistical analysis was performed using one-way ANOVA with Dunnett’s post hoc test (vs. untreated). (n = 3 biological replicates per group) (b) Label-free quantification (LFQ) of the mutant TTR peptide (AADAWEPFASGK), corresponding to the IFM#4 allele, detected in serum by nanoLC-MS/MS. The mutant peptide was detected exclusively in the Cas9-treated group. Error bars represent mean ± SEM. Structural and biochemical comparison of the IFM#4 TTR variant (c-i). (c) Superimposed 3D structures of wild-type (WT; PDB: 3i9p, green), V30M mutant (PDB: 3 kgs, orange), and IFM#4 mutant (predicted by AlphaFold2, magenta). (d–g) Close-up views of amino acid side chains surrounding the mutation site in (d) WT, (e) IFM#4, and (f) V30M structures. (g) The location of the mutation is indicated by square dots in the tetrameric TTR structure. IFM#4 alters local β-sheet structures in the TTR monomer. (h) Aggregation propensity of WT, V30M, and IFM#4 TTR proteins at pH 4.5 and 7.0, assessed by turbidity measurements. IFM#4 peptide showed enhanced aggregation at pH 4.5, comparable to the V30M mutant. (i) SDS-PAGE analysis of TTR samples incubated under acidic conditions (pH 4.5–7.0). Bands corresponding to TTR monomers ( ~ 25 kDa), dimers ( ~ 36 kDa), and tetramers ( ~ 55 kDa) are indicated. IFM#4 and V30M failed to form tetramers, unlike wild-type TTR.
Extended Data Fig. 7 Detection of in-frame mutations (IFMs) in Cas9-edited MEF cells from exon-humanized TTR mice.
(a) Relative genome editing efficiency in MEF cells transfected with mRNA encoding Cas9/NTLA2001 or Cas3/crRNA#2, assessed by ddPCR using the hTTR-ex2 probe. (n = 3 independent biological replicates (separately seeded and transfected MEF cell cultures)) (b) Amino acid sequences of in-frame mutations (IFMs) identified from amplicon sequencing of Cas9-edited MEF cells. IFM#4 comprising ~10% of edited alleles. (c) Alignment of amplicon sequence reads highlighting IFMs in Cas9-treated MEF cells. Error bars represent mean ± SEM. Statistical significance was assessed by a two-tailed Student’s t-test.
Extended Data Fig. 8 Histological analysis of cardiac tissue from TTR exon-humanized mice.
(a) DFS staining of paraffin-embedded heart sections from 9-month-old TTR exon-humanized mice. No overt cardiac TTR amyloid deposition was detected. (b, c) Immunofluorescence staining of frozen heart sections from 2- and 9-month-old TTR exon-humanized mice using anti-TTR and anti-CD68 antibodies. TTR deposition accompanied by CD68⁺ macrophage accumulation was observed in cardiac tissue. (d) Quantification of TTR⁺ and CD68⁺ areas, as well as colocalized regions (CD68/TTR), showed increasing signals from 2 months of age, with a marked elevation around 9 months. (n = 5 biological replicates per group) Error bars represent the mean ± SEM. Statistical analysis was performed using a two-tailed Student’s t-test or a two-tailed Welch’s t-test (for CD68/TTR). (e, f) Frozen heart sections from 8-month-old TTR exon-humanized mice, either untreated (e) or treated with DiT-Cas3-LNP (4.6 mg/kg) (f), were stained with anti-TTR and anti-CD68 antibodies to assess TTR deposition and macrophage infiltration.
Extended Data Fig. 9 On-target editing profile of CRISPR-Cas3 in human HepG2 cells.
(a) Deletion patterns in HepG2 cells transfected with pCas3 carrying the UT-05 crRNA were analyzed by short-read whole-genome sequencing (WGS). Cas3-induced deletions displayed a predominantly processive profile, consistent with results from mouse Hepa1-6 cells (Fig. 2). (b) Quantitative assessment of B4GALT6 exon 9 deletions following plasmid- or mRNA-based delivery of Cascade-Cas3 in HepG2 cells. Droplet digital PCR (ddPCR) analysis was performed to evaluate deletion frequencies at the hB4GALT6 exon 9 locus. Plasmid-based delivery of Cascade-Cas3 induced a 13.71% deletion, whereas mRNA-based delivery resulted in a substantially lower deletion frequency of 5.02%. For comparison, Cas9-NTLA2001 delivered via px458 plasmid induced a 4.93% deletion at the same locus. Similarly, in the ddPCR analysis using a probe located between the TTR gene and its upstream neighboring gene Desmoglein-2 (DSG2)—specifically, approximately 35 kb upstream of TTR Exon 1 and 7 kb downstream of DSG2 Exon 15—the mRNA-based delivery of Cas3 induced only 3.40% modification, comparable to the 2.25% observed with Cas9/NTLA-2001 delivered via px458. Bars represent mean values, and dots represent individual data points (n = 2 independent biological replicates (separately seeded and transfected HepG2 cell cultures). Data are shown as mean ± SEM.
Extended Data Fig. 10 Genome-wide specificity analysis of CRISPR–Cas3 and Cas9 editing in human HepG2 cells using short- and long-read whole-genome sequencing.
(a) The number of predicted potential off-target (POT) sites identified using GGGenome for Cas3-crRNA. (b) Summary of short-read whole-genome sequencing (WGS) coverage and mapping statistics for HepG2 cells transfected with pCas3 carrying the UT-05 crRNA, and for untransfected controls. The average sequencing depth was 139.4×, providing high sensitivity for structural variant detection. (c) A total of 3,035 candidate deletion regions with a split-read ratio ≥6.44 (corresponding to estimated editing efficiency ≥4%) were identified. Among these, 15 overlapped with POT sites. (d) Genome-wide distribution of candidate deletion events revealed a single prominent peak corresponding to the on-target site, with no other significant signals across the genome. (e) IGV snapshot of a representative candidate region among the 15 overlapping POT sites. All observed split-read signals were confined within <1 kb and mapped to low-complexity or repetitive genomic regions. No reproducible structural variants were detected across biological replicates. (f) Oxford Nanopore long-read WGS in Cas3-crRNA UT05 and Cas9-gRNA NTLA2001 transfected HepG2 cells. (g) Cas3-mediated deletions were restricted to the on-target locus under the tested conditions, with no detectable off-target large deletions elsewhere in the genome. Off-target mutations identified in Cas9-edited HepG2 cells using whole-genome sequencing (h-i). (h) Summary of predicted potential off-target (POT) sites for Cas9-gRNA NTLA2001 as identified by Cas-OFFinder. (i) Detection of the on-target deletion at the human TTR locus by short-read whole-genome sequencing (WGS). (j) Among the computationally predicted off-target sites, three loci—POT93, POT200 (both intergenic), and POT88 (intron of PLCB4)—showed measurable deletion signals. The table summarizes chromosomal location, strand orientation, locus annotation, and deletion frequency relative to the on-target site. These off-target events occurred at lower frequencies than the on-target editing and were confined to noncoding or intronic regions.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–3.
Supplementary Tables (download XLSX )
Supplementary Table 1: Summary of in vivo genome-editing clinical trials. Supplementary Table 2: Sequence of crRNAs target in mouse Ttr gene. Supplementary Table 3: List of potential off-target sites for Cas3. Supplementary Table 4: List of potential off-target sites for Cas9. Supplementary Table 5: Sequence of crRNAs target in human TTR gene. Supplementary Table 6: Primer sequences for targeted region. Supplementary Table 7: Sequences of ddPCR assay primers and probes. Supplementary Table 8: Primer sequences for RT–qPCR. Supplementary Table 9: Primer sequences for amplicon sequencing.
Supplementary Data 1
Complete GenBank format sequence of the pCas3 plasmid used for CRISPR–Cas3-based genome editing.
Source data
Source Data Figs. 1 and 4 (download TIF )
Unprocessed images of all gels and blots.
Source Data Extended Data Figs. 4–6 (download TIF )
Unprocessed images of all gels and blots.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ishida, S., Sato, Y., Chosa, K. et al. CRISPR–Cas3-based editing for targeted deletions in a mouse model of transthyretin amyloidosis. Nat Biotechnol (2026). https://doi.org/10.1038/s41587-025-02949-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41587-025-02949-6
