
Abstract
R-loops are DNA–RNA hybrid structures that may promote mutagenesis. However, their contribution to human Mendelian disorders is unexplored. Here we show excess de novo variants in genomic regions that form R-loops (henceforth, ‘R-loop regions’) and demonstrate enrichment of R-loop region variants (RRVs) in ribozyme, snoRNA and snRNA genes, specifically in rare disease cohorts. Using this insight, we report neurodevelopmental disorders (NDDs) caused by rare variants in two major spliceosomal RNA encoding genes, RNU2-2 and RNU5B-1. These, along with the recently described RNU4-2-related ReNU syndrome, provide a genetic explanation for a substantial proportion of individuals with NDDs.
Similar content being viewed by others


Main
R-loops form predominantly at sites of active transcription and may promote mutagenesis through exposure of single-stranded DNA to cytidine deaminases, nucleases, genotoxins or transcription–replication conflicts (Fig. 1a and Extended Data Fig. 1)1,2,3. Several methods for identifying R-loops exist4. By intersecting consensus R-loop regions (genomic footprint of 4.32%)5 with 975,406 variants in the 100,000 Genomes Project (100KGP)6 rare disease de novo (DN) dataset, we found 53,116 (5.4%, median = 4 per trio, range = 1–46) to be in R-loop regions. In regions that are well covered in gnomAD, we detected substantial excess of DN variants in R-loop versus non-R-loop regions of promoters, exons, introns and random genomic sequences (Fig. 1b). To test whether this observation could be due to R-loop regions having inherently higher sequence-context mutability, we ascertained all bioinformatically predicted R-loop-forming sequences and found the DN variant rate to be substantially higher in experimentally validated regions than in regions without experimental evidence, although the average mutability of the two groups was not significantly different (Extended Data Fig. 2 and Supplementary Table 1)7. All these findings, apart from excess DN RRVs in promoters, were validated in an independent 1,548 healthy Icelandic trios genome sequencing dataset (Extended Data Fig. 3)8. These results suggest that DN variants are more common in R-loop regions.

a, Schematic of R-loops and the potential mutagenic processes that could result in increased mutagenesis in these regions. b, Violin plots showing mutation rate in 100KGP for genomic features that overlap (RL) or do not overlap (noRL) experimentally determined R-loop regions. We randomly selected 500 subset regions from each group for 1,000 iterations (***P < 2.22 × 10−16, two-sided Wilcoxon test). Violins extend from minimal to maximal data points. Box plots are centered on median with interquartile ranges as outer bounds, error bars as s.e.m and outliers as dots. c, DN RRV enrichment dot plot in GENCODE noncoding biotypes in 100KGP and Iceland control cohort. The red line marks the log2 fold enrichment threshold. d, Bubble plot of DN RRV enriched gene biotypes in 100KGP. e,f, Gene diagrams of RNU2-2 (e) and RNU5B-1 (f) with variant depletion as heatmap derived from gnomADv4. Variants are color coded by cohort of origin, with filled circles denoting DN variants and gradient-filled circles denoting unknown inheritance or parental transmission. Statistical data underlying the plots are provided as source data.
Next, we explored whether the distribution of DN RRVs is different in disease and control cohorts. We found significant genomic footprint-adjusted >2 log2 fold enrichment for DN RRVs exclusively in the rare disease cohort only for ribozyme, snoRNA and snRNA gene biotypes (Fig. 1c and Supplementary Table 2). These groups comprised of three distinct variants in two ribozyme genes, 15 variants in 12 snoRNA genes, and 86 variants in 18 snRNA genes (Fig. 1d and Supplementary Table 3), including five known recessive disease genes (Supplementary Table 4) and dominant ReNU syndrome (OMIM 620851) associated RNU4-2 (n = 36)9,10. These results validated our approach to detect disease-causing variants.
We next focused on genes without known disease associations and two or more new (absent from gnomADv4) DN RRVs in the rare disease cohort—RNU2-2 (formerly RNU2-2P; five variants in nine individuals) and RNU5B-1 (four variants in four individuals; Extended Data Fig. 4). The 100KGP DN variant dataset was generated with high stringency such that some real variants passing the base filter (based on read depth and zygosity in each sample) are excluded as they may fail on one of the stringency filters. For the two genes of interest, therefore, we released the stringency filter and identified three additional individuals with DN RNU2-2 or RNU5B-1 variants (Extended Data Fig. 5). These data indicated that RNU2-2 and RNU5B-1 could be new disease genes.
We next investigated the distribution of DN RRVs detected in RNU2-2 and RNU5B-1 in the rare disease cohort and found that they occurred in regions constrained for variants in gnomADv4 (n = 76,215 individuals) and were absent from individuals coded as ‘unaffected’ (n = 32,030 participants) in 100KGP. This included five DN variants in a constrained region of RNU2-2 (from 1 to 60 bp; ENST00000410396.1:n.3C>A, n.4G>A, n.7_8insA, n.35A>C and n.35A>G) in nine individuals (Fig. 1e). Notably, RNU2-2 shares high sequence similarity with RNU2-1 (Extended Data Fig. 6). RNU2-1 resides in a 6.1-kb tandem repeat on chromosome 17 (ref. 11). This locus was not annotated in GRCh37 but is included in GRCh38 with thirteen repeat units. Importantly, eight nucleotides differ between RNU2-2 and RNU2-1, allowing for unambiguous read alignment in GRCh38. For RNU5B-1, we found three DN variants in a constrained region (from 30 to 50 bp; ENST00000363286.1:n.37G>C, n.42_43insA and n.44A>G) in four individuals (Fig. 1f). Two individuals had new DN RNU5B-1 variants outside of this constrained region, one of which (n.100C>G) had an alternative diagnosis that explained their phenotype. The other (n.59G>C) was an unsolved individual; however, this variant occurs in three other unaffected individuals in 100KGP. These data identify constrained regions in RNU2-2 and RNU5B-1 that may harbor disease-causing variants.
Next, we set out to expand the RNU2-2 and RNU5B-1 cohorts. Within the 100KGP database, we identified six other individuals with one of the abovementioned five RNU2-2 heterozygous variants that were not DN or could not be proven to be DN (parental samples not available in four cases, and one instance each of maternal and paternal transmission). Expanding the search for other variants within the n.30–n.50 region of RNU5B-1 identified one further individual in 100KGP with n.39_40insT, the DN origin of which was determined through phasing of maternal SNPs in this duo sequencing set up (Extended Data Fig. 7). Analysis of the Genomics England, National Genomics Research Library (NGRL; n = 32,203 genomes, 6,354 trios), SolveRD12,13 (n = 2,859 whole-genome sequencing (WGS)), the Centre for Population Genomics (Australia, n = 4,704 WGS, 864 trios) and South Korean Undiagnosed Diseases (n = 1,089 WGS probands) databases identified ten more individuals with rare RNU2-2 variants and four with rare RNU5B-1 variants in the constrained regions, bringing the total to 27 individuals for rare variants in RNU2-2 (Fig. 1e) and nine for RNU5B-1 (Fig. 1f).
Next, we studied phenotypes of the RNU2-2 and RNU5B-1 cohorts. We found that all 21 probands with rare RNU2-2 or RNU5B-1 variants in 100KGP were previously genetically unsolved. Human Phenotype Ontology (HPO) terms including severe global developmental delay (odds ratio (OR) = 33.6, P < 10−8), generalized hypotonia (OR = 15.8, P = 1.2 × 10−8) and microcephaly (OR = 9.0, P = 1.1 × 10−5) were significantly enriched in individuals with RNU2-2 variants in comparison with other rare disease probands (Fig. 2a). We were able to collect detailed clinical information from seven individuals with rare RNU2-2 variants in our cohort, which revealed that the ‘RNU2-2-related disorder’ is characterized by global developmental delay, prominent speech impairment, epilepsy and, in some cases, presentation reminiscent of Pitt–Hopkins or Rett syndrome (Fig. 2b, Supplementary Tables 5 and 6 and Supplementary Note). Generalized hypotonia (OR = 37.3, P = 7.4 × 10−5), macrocephaly (OR = 30.6, P = 1.8 × 10−4), failure to thrive (OR = 24.7, P = 4.5 × 10−4), global developmental delay (OR = 22.1, P = 5.6 × 10−3) and abnormality of the eye (OR = 18.7, P = 1.4 × 10−3) were significantly enriched in individuals with RNU5B-1 variants (Fig. 2c). We collected detailed clinical information from five individuals with rare RNU5B-1 variants in our cohort, which revealed that the ‘RNU5B-1-related disorder’ is characterized by global developmental delay, relative macrocephaly, seizures and failure to thrive (Fig. 2d, Supplementary Tables 7 and 8 and Supplementary Note).

a,b, ORs for HPO terms in RNU2-2 (a) and RNU5B-1 (b) cases compared to all probands in the rare disease arm of 100KGP. Only HPO terms observed in at least three RNU2-2 cases (*P < 0.0045, two-sided Z test of the log OR) or at least two RNU5B-1 cases (*P < 0.0071, two-sided Z test of the log OR) are shown (exact P values are provided in Supplementary Table 10). c,d, Facial photographs and MRI images of affected individuals with RNU2-2 (a) and RNU5B-1 (b) variants. MRI brain of Individual 2 shows cerebral and cerebellar parenchymal volume loss. MRI brain of Individual 5 shows hypoplastic corpus callosum. Written informed consent for each individual was obtained from families for publication in this paper.
As the predominant phenotypes of the two syndromes are neurodevelopmental, we asked whether the two genes are expressed in the developing human brain. As snRNAs are not polyadenylated, we used small RNA-seq data from ENCODE human developing brain and a related tissue, retina. We used a stringent bioinformatic protocol to remove multimapping reads as a confounder by including only primary alignments. Both datasets showed high expression of RNU2-2 (higher than its multicopy paralog, RNU2-1) and RNU5B-1 (Fig. 3a,b). Notably, RNU2-2 was annotated as a pseudogene during the period of this study but encodes U2-2, a U2 small nuclear RNA that has previously been shown to be incorporated into the spliceosome, although its role remains putative14,15. Our small RNA-seq analysis shows that RNU2-2 is highly expressed and not a pseudogene.

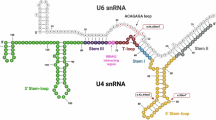
a, Balloon plot of small RNA-seq expression data with stringent multimapping protocol for RNU2, RNU4 and RNU5 paralogs in human developing brain derived from ENCODE. Normalized expression is in primary alignments per million. b, Box plots of small RNA-seq expression data with stringent multimapping protocol for RNU2, RNU4 and RNU6 paralogs in the human choroid (n = 13), neurosensory retina (NSR, n = 4) and retinal pigment epithelium (RPE, n = 16). Data are represented in box plots and the median value is central. c, Schematic representation of RNU2-2 variants mapped to the U2–U6 structure in complex with the pre-mRNA branch point. d, Schematic representation of RNU5B-1 variants mapped to the U5 structure in complex with the acceptor and donor sites of adjacent exons, amended from ref. 20.
Next, we explored the effects of the six RNU2-2 variants identified in the study. All variants are transversions or insertions affecting positions n.3, n.4, n.5 and n.7 located within the 5' end of U2 and predicted to disrupt interaction with the 3' end of U6 in the major spliceosome (Fig. 3c)16. The n.35A nucleotide is in the branch site recognition sequence (GUAGUA) that base-pairs with the conserved U of the pre-mRNA branch point (YNYURAY, where Y, pyrimidine; N, any nucleotide; and R, purine)17. A dominant-negative effect of variants at the yeast U2 36A (orthologous to human n.35A) position is supported by functional analysis in vitro that found the U2 36A>G variant does not support splicing18. Importantly, although none of the variants in our cohort are present in the general population, different heterozygous variants (n.3C>T and n.7_8insG) at the same positions have been reported in gnomADv4, suggesting that only specific variants in the constrained regions of RNU2-2 may be pathogenic or that some may demonstrate reduced penetrance.
We also studied the effects of the five RNU5B-1 variants identified in the study. RNU5B-1 encodes U5B-1, one of five U5 paralogs in the human genome (Extended Data Fig. 8). The RNU5B-1 variant positions lie within the invariantly conserved loop I structure of U5, essential for holding and aligning the two exons for accurate joining during splicing (Fig. 3d)19,20. Notably, a different substitution in the constrained region, n.39C>T, is observed in three individuals in gnomADv4.
In summary, we show that DN variants are frequent in R-loop regions and that variants in snRNA components of the major spliceosome show enrichment in a disease cohort in comparison with a control cohort. We implicate rare variants in RNU2-2 and RNU5B-1 as causes for neurodevelopmental disorders (NDDs). RNU2-2-related disorder is characterized by global developmental delay, intellectual disability, microcephaly, autistic behavior and tendency for seizures. RNU5B-1-related disorder is characterized by global developmental delay, hypotonia, macrocephaly and failure to thrive. Along with variants in RNU4-2, variants in RNU2-2 and RNU5B-1 provide a genetic explanation for an exceptionally large proportion of individuals with NDDs caused by variants in nonprotein-coding genes and previously unsolved genetic NDDs (Supplementary Table 9). Our approach also demonstrates the utility of incorporating information about DNA secondary structures in variant analysis for identifying noncoding transcribed regions.
Methods
The research presented here complies with all relevant ethical regulations and was performed under the approvals given by the South Manchester National Health Service (NHS) Research Ethics Committee (REC; 11/H1003/3/AM02), Cambridge South NHS REC (14/EE/1112–100KGP), University of Tübingen (ClinicalTrials.gov registration: NCT03491280–SolveRD), Seoul National University Hospital Institutional Review Board (2006-059-1131 and 2407-195-1559) and the Royal Children’s Hospital Human Research Ethics Committee (HREC/67401/RCHM-2020–Rare Diseases Now). Informed consent was obtained from all participants or their legal guardians.
Genomic distribution of R-loop regions
To investigate the genomic distribution of R-loop regions, CHIPSeeker was used through the Galaxy platform to annotate R-loop regions based on their genomic context. GENCODE v.32 was used as the input gtf file.
Cohort information
100KGP
Participants were recruited to 100KGP with diverse rare disease phenotypes. Information about 100KGP is available at https://www.genomicsengland.co.uk/initiatives/100000-genomes-project and the preliminary report6. The rare disease cohort was derived from 73,527 genomes (release v19). The DN dataset was derived from 13,949 rare disease trios (active consent was available from 12,565 trios at the time of this study). The group of unaffected individuals in 100KGP was defined by keeping individuals with ‘unaffected’ as their status in the participant_summary table in LabKey.
NGRL
The NHS Genomic Medicine Service (https://re-docs.genomicsengland.co.uk/gms_release4/) data (30,047 rare disease genomes, including 15,889 from probands) in the NGRL was searched in the Genomics England Research Environment (GERE). Detailed information on indications for WGS in the NHS can be found at https://www.england.nhs.uk/publication/national-genomic-test-directories/. Of 15,889 probands, 6,321 (39.7%) were recruited for R27 (multiple congenital malformations and dysmorphic syndromes) or R29 (intellectual disability).
SolveRD
SolveRD sequencing data were accessed through the RD-CONNECT platform (https://rd-connect.eu/). SolveRD includes 523 genomes and 9,351 exomes from 9,645 individuals. Disease categories for the recruitment to SolveRD comprise rare neurological diseases (n = 2,271 families), (multiple) malformation syndromes, intellectual disability and other neurodevelopmental disorders (ITHACA and SpainUDP, n = 1,857), rare neuromuscular diseases (EURO-NMD, n = 1,517) and suspected hereditary gastric and bowel cancer (GENTURIS, n = 359). Only WGS datasets were included in this analysis, for which 334 correspond to affected individuals21. The proportion of each recruitment category with WGS is not available.
Australia
The Centre for Population Genomics hosts sequencing data for Australian individuals suspected of living with rare genetic conditions. Neurodevelopmental disorders are a major focus, although other key disease areas include epileptic encephalopathies, leukodystrophies, mitochondrial and renal conditions. As of this analysis, the dataset contains WGS data for 2,845 affected Individuals from 2,564 families (total Individuals with WGS data = 4,704). In 864 cases, full parental trio WGS was available22.
South Korea
A total of 3,976 participants were recruited from the Korea BioBank and a pediatric rare disease cohort comprising patients and their relatives from SNUH and its network hospitals. Among these, 1,150 individuals presented with NDDs, with or without epilepsy, that remained genetically undiagnosed despite prior microarray and exome sequencing analyses. To refine the cohort, kinship analysis was conducted to identify unrelated individuals, resulting in a final study cohort of 1,089 probands23.
Identification and characterization of DN variants within R-loop regions in the 100KGP
The project was registered with the Genomics England research registry (RR1147) and received approval to access data from 100KGP (Genomics England, NGRL v.5.1: https://doi.org/10.6084/m9.figshare.4530893/v7).
Chromosomal coordinates, aligned to GRCh38, were downloaded from previously published consensus R-loop regions (RL regions) derived from 810 R-loop mapping datasets in humans5. These were converted to BED format and imported to GERE using the Airlock.
This BED file was intersected with a BED file generated from the DN variant dataset in GERE (main_programme_v18_2023-12-21) using the bedtools24 intersect function, with default parameters. To generate the de novo BED file, GRCh37 coordinates in the GERE were lifted over to GRCh38 using UCSC-liftOver, and then, the start position of each variant in the harmonized file was taken as the start position in the BED file and the end position was ‘start +1’. For initial analysis, only high-confidence DN variants in GERE flagged by a stringency criterion of ‘1’ were included. DN variants that failed the stringency filter were included for genes of interest and sequencing reads were manually inspected in IGV. Further information on the DN variant dataset in 100KGP can be found at https://re-docs.genomicsengland.co.uk/de_novo_data/. The DN dataset was generated with two quality filters—base and stringent. The base filter is applied on zygosity (heterozygous in the offspring and homozygous reference in the parents) and read depth (minimum 20× and maximum 98×) in all samples of the trio. If the variant fails any of these filters, it fails the base filter. Variants that pass base filter are then subject to the stringent filter, which include the following: altreadparent_filter (no more than one read supporting the alternate allele in either the mother or the father); abratio_filter (the AB ratio in the offspring is between 0.3 and 0.7); proximity_filter (DNV is not located within 20 bp of another DNV within the same trio); segmentalduplication_filter (no overlap with segmental duplications); simplerepeat_filter (no overlap with simple repeat regions; patch_filter (no overlap with patch regions); stringent_filter (**all base and stringent filters pass). Variants that failed the stringency filter in RNU2-2 (a single instance of n.7_8insA) and RNU5B-1 (two instances of n.42_43insA) failed due to the ‘altreadparent’ filter, although no good quality alternate reads could be identified in the sequencing data in IGV.
All data were exported through the Airlock for preparation of this paper.
Analysis of DNMs occurring in the Icelandic control dataset versus 100KGP
The DNM file for the genome sequencing of 1,548 trios in the Icelandic study8 was downloaded and used to generate a BED file of genomic coordinates (GRCh38). This file was intersected with the GENCODE v32 GTF file to extract overlaps with genes and then the number of overlaps for each gene type in GENCODE was counted. We repeated this analysis for DNMs in 100KGP. We used the genomic footprint of each gene type to calculate the number of DNMs per bp per trio for each group. We then calculated the ratio of DNMs comparing 100KGP to Icelandic controls. For instance, if a biotype has a proportional footprint on 1% of the human genome in GRCh38, then 1% of DN variants would be expected to occur in this biotype, assuming a uniform distribution of DN variants. If this biotype actually contains 10% of all DN variants, then it is enriched tenfold compared to genomic footprint expectation. The expected number of DN RRVs was used as the probability for the binomial test, observed values as the success and the total number of DN RRVs as the trials. Adjustment for multiple testing was made by multiplying the P value by the number of GENCODE biotypes. Fold change was calculated as the number of observed DN RRVs divided by the expected number of RRVs for each biotype. Plots were generated in R using ggplot2 package. All source data are provided.
Analysis of DNM enrichment in R-loop regions
A total of 50,000 random 1,000-bp genomic regions were generated from GRCh38 using bedtools random function. To generate regions with comparable selective pressure to R-loop regions, BED files were generated for all promoters and exons annotated by Ensembl regulatory build (GRCh38, 2016-11-11 and GENCODE v32, respectively). A BED file for GRCh38 introns was generated by subtracting the exon coordinates and overlapping with gene coordinates. Regions overlapping R-loop regions were identified using bedtools intersect and either removed with ‘-v’ option to generated ‘noRL’ BED files or selected to generate ‘RL’ BED files.
For each BED file, 500 random entries were selected (using shuf *.bed | head -n 500). These random sets of regions were separately intersected with 100KGP and Iceland DNM BED files. For each entry in the BED file, the number of overlapping DNMs was normalized to the entry genomic length, and then the sums of all regions were calculated and divided by the number of trios to give the total number of DNMs per bp per trio. The randomization was performed 1,000 times for each group—exons, promoters, introns and random. Results were plotted in R using ggplot2 as a grouped violin plot, and the ggsignif R package was used to calculate the Wilcoxon test P values between noRL and RL groups for each feature.
R-loop forming sequence analysis
R-loop forming sequences (RLFSs) for GRCh38 were predicted by QmRLFS-finder25 and downloaded from the UCSC browser session provided in ref. 5. RLFSs were split into two groups—RLFS_RL, which overlap R-loop consensus regions and RLFS_noRL, which do not overlap consensus R-loop regions. This intersection was achieved using bedtools intersect. The subsampling procedure described above for genomic features was performed to calculate the mutation rate (DN variants per bp per generation). Statistical significance was calculated using the Wilcoxon test.
The mutability of RLFS_RL and RLFS_noRL was compared by extracting the Roulette annotations from the VCFs provided at http://genetics.bwh.harvard.edu/downloads/Vova/Roulette/. Where available, the adjusted mutation rate (AR) was extracted; otherwise, the raw score (MR) was used. The Roulette mutation rates for each possible SNV at each position within each RLFS were summed. The summed mutation rates were divided by the total number of all possible SNVs within each group to calculate the mean mutability per SNV for RLFS_RL and RLFS_noRL.
Analysis of gnomADv4 coverage data
As coverage by WGS is reduced at repetitive regions, which may be annotated GENCODE biotypes or RLFS, we sought to identify regions of GRCh38 that were poorly covered in gnomADv4 to remove them from downstream analysis. We used the coverage data from gnomADv4 to select all nucleotides where fewer than 50% of individuals had 20× coverage. We then merged these into regions using bedtools merge with a flank of 20 bp, meaning that all poorly covered nucleotides within 20 bp of each other would be merged into a single region. We then removed these regions from downstream analysis using bedtools intersect -v.
Analysis of variant depletion in gnomADv4
Due to the high mutability and strong selection pressures, the sliding window-based strategy9 was used to identify variant-depleted regions in the two genes. All PASS single-nucleotide substitutions in RNU2-2 and RNU5B-1 present in gnomADv4 were extracted using bcftools view -v snps *.vcf.gz | bcftools query -f ‘%CHROM %POS %REF %ALT\n’. A BED file with an entry representing each distinct variation in gnomADv4 was generated. Bedtools makewindows was used to generate 10-bp nonoverlapping windows for RNU2-2 and RNU5B-1. Each gene window BED file was intersected with the gnomADv4 variant BED file and the number of overlaps for each window was counted. The number of possible substitutions was calculated for each window (nominally 30, for a 10-bp window, as each nucleotide has three possible substitutions). The number of observed substitutions in gnomADv4 was then divided by the number of possible substitutions. The resultant value was then normalized to the median of the window, meaning that strongly depleted regions have negative values, while variant-tolerant regions have positive values. Variant-depleted region was defined as one that had a normalized observed proportion of SNVs of less than −0.2.
Analysis of gene snRNA expression in developing human brain using small RNA-seq
BAM files for small RNA-seq experiments in human brain, following treatment with tobacco acid phosphatase, were downloaded from ENCODE and aligned to GRCh38. Accession numbers for experiments used were ENCSR000AFR (diencephalon), ENCSR000AFY (parietal), ENCSR000AFX (occipital), ENCSR000AFS (frontal), ENCSR000AGD (temporal) and ENCSR000AFQ (cerebellum).
For each BAM file (two per tissue, representing two biological replicates), the total number of primary alignments for each gene was extracted using samtools view (‘samtools view -L gene.bed -c -F 260 $bam’). The total number of primary alignments in the BAM file (‘samtools view -c -F 260 $bam’) was then used to generate a normalized expression value by dividing the gene of interest primary alignment by all alignments, divided by 1 million (resulting in primary alignments per million). Data were plotted as balloon plots using ggplot2 in R.
Analysis of gene snRNA expression in human retina
RNA was isolated from human donor eye tissue, which was collected and dissected as described previously26 from an ethically approved Research Tissue Bank (UK NHS Health Research Authority reference: 15/NW/0932). Total RNA was isolated from four neurosensory retina samples, 16 pelleted retinal pigment epithelium samples and 13 choroid samples that had been stored in RNAlater, using an Animal Tissue RNA Purification Kit (Norgen Biotek), as per the manufacturer’s instructions. Sequencing libraries were prepared using the NEBNext Multiplex Small RNA Library Prep Kit, as per the manufacturer’s protocols, with size selection performed using AMPure XP beads. Paired-end sequencing (2 × 75 bp) was performed on an Illumina HiSeq 4000.
NEBNext adapters were removed from sequencing reads using trimmomatic before alignment against the GRCh38 reference genome with bowtie. No mismatches between sequencing reads and the reference genome were allowed, and no restriction was set on multimapping reads. Sequence read counts were restricted to primary alignments using samtools v1.9, and therefore only counted once if they aligned to multiple RNU2 or RNU5 genes or pseudogenes. Calculations were drawn from read1 datasets and normalized for the total read count achieved for the sample.
Multiple sequence alignment
Genomic fasta sequences for U2 and U5 paralogs were downloaded from Ensembl. Fasta sequences for RNU2-1 (ENSG00000274585), RNU2-2 (ENSG00000222328), RNU5A-1 (ENSG00000199568), RNU5B-1 (ENSG00000200156), RNU5D-1 (ENSG00000200169), RNU5E-1 (ENSG00000199347) and RNU5F-1 (ENSG00000199377) were used. Alignments were performed using the Clustal Omega web tool and imported into MView for visualization. The percent identity matrix for U5 was imported into RStudio and the heatmap was plotted using the pheatmap package.
Phenotype matching and analysis
Phenotypic information for individuals carrying candidate variants was extracted from the LabKey rare disease phenotype database (main_programme_v18_2023-12-21) using HPO terms. The analysis focused on identifying common phenotypes across individuals sharing these variants. For individuals with RNU2-2 and RNU5B-1 variants, the occurrence of each HPO term was counted, and ORs were calculated for term against all other individuals (n = 39,755) in the cohort. Significance thresholds were adjusted by Bonferroni correction.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Genomic and phenotypic data are available for the 100KGP and individuals who have had WGS through the Genomic Medicine Service in the NGRL. Access to the NGRL may be granted following application via https://www.genomicsengland.co.uk/research/academic/join-research-network, which gives access to the secure GERE. Genomic data used pertain to participants in 100KGP in the Main Programme v.18 and the GMS data v.4. SolveRD data are accessible by application through the RD-CONNECT platform. Data presented in this paper were requested for the Airlock transfer on 26 September 2024. The paper was submitted for approval by the Genomics England Publication Committee on 27 September 2024 and was approved on 3 October 2024. Access to the Australian Centre for Population Genomics and South Korean Undiagnosed Diseases Program datasets can be requested through contact with the authors. The GRCh38 human genome reference assembly can be accessed at https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001405.26/. The GENCODE v.32 comprehensive annotations were accessed within the GERE but can be downloaded from https://www.gencodegenes.org/human/release_32.html. The ENCODE data can be accessed at https://www.encodeproject.org/ and relevant accession codes are provided in Methods. The gnomADv4 genotype VCF files were accessed within the GERE but can also be downloaded from https://gnomad.broadinstitute.org/. Small RNA-seq datasets analyzed in this study are available at the NCBI Sequence Read Archive through accession PRJNA1256119. Source data are provided with this paper.
Code availability
Software packages bedtools (v.2.31.0), bcftools (v.1.16) and samtools (v.1.9) were used in this work. Rv4 with RStudio, ggplot2 (v.3.5.2) and ggsignif (v.0.6.4) was used for plotting. Code generated for variant analysis within GERE is located within the secure GERE and available upon request.
Change history
24 June 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41588-025-02274-3
References
Belotserkovskii, B. P., Tornaletti, S., D'Souza, A. D. & Hanawalt, P. C. R-loop generation during transcription: formation, processing and cellular outcomes. DNA Repair 71, 69–81 (2018).
García-Muse, T. & Aguilera, A. R loops: from physiological to pathological roles. Cell 179, 604–618 (2019).
McCann, J. L. et al. APOBEC3B regulates R-loops and promotes transcription-associated mutagenesis in cancer. Nat. Genet. 55, 1721–1734 (2023).
Yan, Q. et al. Proximity labeling identifies a repertoire of site-specific R-loop modulators. Nat. Commun. 13, 53 (2022).
Miller, H. E. et al. Quality-controlled R-loop meta-analysis reveals the characteristics of R-loop consensus regions. Nucleic Acids Res. 50, 7260–7286 (2022).
The 100,000 Genomes Project Pilot Investigators. 100,000 Genomes pilot on rare-disease diagnosis in health care—preliminary report. N. Engl. J. Med. 385, 1868–1880 (2021).
Seplyarskiy, V. et al. A mutation rate model at the basepair resolution identifies the mutagenic effect of polymerase III transcription. Nat. Genet. 55, 2235–2242 (2023).
Jónsson, H. et al. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature 549, 519–522 (2017).
Chen, Y. et al. De novo variants in the RNU4-2 snRNA cause a frequent neurodevelopmental syndrome. Nature 632, 832–840 (2024).
Greene, D. et al. Mutations in the U4 snRNA gene RNU4-2 cause one of the most prevalent monogenic neurodevelopmental disorders. Nat. Med. 30, 2165–2169 (2024).
Tessereau, C. et al. Estimation of the RNU2 macrosatellite mutation rate by BRCA1 mutation tracing. Nucleic Acids Res. 42, 9121–9130 (2014).
Matalonga, L. et al. Solving patients with rare diseases through programmatic reanalysis of genome–phenome data. Eur. J. Hum. Genet. 29, 1337–1347 (2021).
Zurek, B. et al. Solve-RD: systematic pan-European data sharing and collaborative analysis to solve rare diseases. Eur. J. Hum. Genet. 29, 1325–1331 (2021).
Mabin, J. W., Lewis, P. W., Brow, D. A. & Dvinge, H. Human spliceosomal snRNA sequence variants generate variant spliceosomes. RNA 27, 1186–1203 (2021).
Bousquets-Muñoz, P. et al. PanCancer analysis of somatic mutations in repetitive regions reveals recurrent mutations in snRNA U2. NPJ Genom. Med. 7, 19 (2022).
Rhode, B. M., Hartmuth, K., Westhof, E. & Lührmann, R. Proximity of conserved U6 and U2 snRNA elements to the 5′ splice site region in activated spliceosomes. EMBO J. 25, 2475–2486 (2006).
Kadri, N. K., Mapel, X. M. & Pausch, H. The intronic branch point sequence is under strong evolutionary constraint in the bovine and human genome. Commun. Biol. 4, 1206 (2021).
McPheeters, D. S. & Abelson, J. Mutational analysis of the yeast U2 snRNA suggests a structural similarity to the catalytic core of group I introns. Cell 71, 819–831 (1992).
McGrail, J. C., Tatum, E. M. & O’Keefe, R. T. Mutation in the U2 snRNA influences exon interactions of U5 snRNA loop 1 during pre‐mRNA splicing. EMBO J. 25, 3813–3822 (2006).
Artemyeva-Isman, O. V. & Porter, A. C. G. U5 snRNA interactions with exons ensure splicing precision. Front. Genet. 12, 676971 (2021).
Laurie, S. et al. Genomic reanalysis of a pan-European rare-disease resource yields new diagnoses. Nat. Med. 31, 478–489 (2025).
Pais, L. S. et al. seqr: a web-based analysis and collaboration tool for rare disease genomics. Hum. Mutat. 43, 698–707 (2022).
Lee, Y. et al. Genomic profiling of 553 uncharacterized neurodevelopment patients reveals a high proportion of recessive pathogenic variant carriers in an outbred population. Sci. Rep. 10, 1413 (2020).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Jenjaroenpun, P., Wongsurawat, T., Yenamandra, S. P. & Kuznetsov, V. A. QmRLFS-finder: a model, web server and stand-alone tool for prediction and analysis of R-loop forming sequences. Nucleic Acids Res. 43, W527–W534 (2015).
Mcharg, S. et al. Mast cell infiltration of the choroid and protease release are early events in age-related macular degeneration associated with genetic risk at both chromosomes 1q32 and 10q26. Proc. Natl Acad. Sci. USA 119, e2118510119 (2022).
Acknowledgements
We thank the participants and the recruiting clinicians of the 100KGP. We thank Genomics England for generating the data and provided the GERE platform for analysis. We thank P. O’Donovan, Z. Mustafa, A.L. Taylor and J. Yang for airlock requests. A.J. and S.B. acknowledge the support of SolveRD. The SolveRD project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement 779257. This study has been delivered through the National Institute for Health and Care Research (NIHR) Manchester Biomedical Research Centre (NIHR203308). S.B. acknowledges the support of the MRC Epigenomics of Rare Diseases (EpiGenRare) Node (MR/Y008170/1). We thank the Macular Society for their support in the generation of retina RNA-seq datasets. A.B. is supported by a Wellcome PhD Training Fellowship for Clinicians and the 4Ward North PhD Programme for Health Professionals (223521/Z/21/Z). Analysis was supported by the Centre for Population Genomics (Garvan Institute of Medical Research and Murdoch Children’s Research Institute) and was funded in part by a National Health and Medical Research Council investigator grant (2009982). This research was made possible through access to data in the National Genomic Research Library, which is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The National Genomic Research Library holds data provided by patients through the NHS as part of their clinical care, as well as data collected during their participation in research. The National Genomic Research Library is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also funded research infrastructure. The views expressed are those of the authors and not necessarily those of the SolveRD, the NIHR or the Department of Health and Social Care.
Author information
Authors and Affiliations
Contributions
N.T., A.J. and A.B. performed analyses. A.J. and S.B. wrote the paper and supervised the project. A.J., S.B., M.R., J. Campbell, N.S., M.B., D.H., A.d.B., T.Y.T., J.H.C., S.Y.K. and S.L. provided clinical information. R.R., C.S., J.H., T.K., and J. Choi performed database searches in respective cohorts. G.R. and S.G.-J. performed the small RNA-seq experiments on retinal samples. J.E. and A.J. performed small RNA-seq analysis in the retina and brain. R.T.O. provided theoretical input. All authors read and approved the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Genomic distribution of consensus experimentally determined R-loop regions.
Pie chart produced by CHIPSeeker showing the distribution of R-loop consensus regions genome-wide.
Extended Data Fig. 2 Rate of de novo variants in 100KGP rare disease cohort in predicted R-loop forming sequences.
The ‘noRL’ group denotes regions that are predicted to form R-loops based on sequence context but lack experimental evidence. The ‘RL’ group denotes regions that are predicted to form R-loops based on sequence context and are supported by experimental evidence. ***P < 0.001 (two-sided Wilcoxon test, n = 1,000). Violins extend from minimal to maximal data points. Box plots are centered on median with interquartile ranges as outer bounds, error bars as s.e.m. and outliers as dots. Mutation rate represents de novo variants per bp per generation. Data underlying this plot are provided as source data.
Extended Data Fig. 3 Rate of de novo variants in Icelandic healthy trio cohort in predicted R-loop forming sequences.
a, Enrichment of de novo variants in RL regions in Icelandic cohort. ***P < 0.001 (two-sided Wilcoxon test, n = 1,000). Violins extend from minimal to maximal data points. Box plots are centered on median with interquartile ranges as outer bounds, error bars as s.e.m. and outliers as dots. Mutation rate represents de novo variants per bp per generation. b, Replication of enrichment for de novo variants in RLFS that overlap consensus R-loop regions. The ‘noRL’ group denotes regions that are predicted to form R-loops based on sequence context, but lack experimental evidence. The ‘RL’ group denotes regions that are predicted to form R-loops based on sequence context and are supported by experimental evidence. ***P < 0.001 (two-sided Wilcoxon test, n = 1,000). Violins extend from minimal to maximal data points. Box plots are centered on median with interquartile ranges as outer bounds, error bars as s.e.m. and outliers as dots. Mutation rate represents de novo variants per bp per generation. Data underlying these plots are provided as source data.
Extended Data Fig. 4 Filter logic to identify new disease genes in this study.
Numbers of variants at each step are shown in lozenge shapes, number of genes in diamond shapes and individuals in parallelograms. For example, the last stage shows the identification of eight variants in two new dominant disorder genes (RNU2-2 and RNU5B-1) in 12 individuals.
Extended Data Fig. 5 De novo variants in R-loop regions affecting genes of interest with absence from gnomADv4.
a, All de novo variants in RNU2-2 identified in 100KGP. b, All de novo variants in RNU5B-1 identified in 100KGP. The bottom lollipops on each plot are variants that failed stringent de novo filtering. Lightly shaded circles indicate the presence of that variant in gnomADv4.
Extended Data Fig. 6 Sequence alignment of RNU2-1 and RNU2-2.
The eight nucleotide differences between RNU2-1 and RNU2-2 are highlighted by red boxes. Nucleotides are colored by identity.
Extended Data Fig. 7 IGV screenshot of mother–daughter sequencing duo for individual #3.
The RNU5B-1 n.39_40insT variant is highlighted with a blue arrow. The informative maternal variant is highlighted by a green arrow. Note that in all the informative reads in the proband, the two variants are present in cis, showing that the proband’s RNU5B-1 n.39_40insT variant is highly likely to have arisen on the maternal allele.
Extended Data Fig. 8 Multiple sequence alignment of U5 paralogs and their sequence identity.
a, Multiple sequence alignment of human U5 paralogs normalized to RNU5F-1, which is the longest sequence. Nucleotides are colored by identity. b, Heatmap of percent identity for U5 paralogs showing that RNU5A-1, RNU5B-1 and RNU5E-1 share highest identity with each other.
Supplementary information
Supplementary Information (download PDF )
Supplementary Note.
Supplementary Tables (download XLSX )
Supplementary Tables 1–10.
Source data
Source Data Fig. 1 (download TXT )
Statistical source data for Fig. 1b.
Source Data Extended Data Fig. 2 (download TXT )
Statistical source data.
Source Data Extended Data Fig. 3 (download XLSX )
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jackson, A., Thaker, N., Blakes, A. et al. Analysis of R-loop forming regions identifies RNU2-2 and RNU5B-1 as neurodevelopmental disorder genes. Nat Genet 57, 1362–1366 (2025). https://doi.org/10.1038/s41588-025-02209-y
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41588-025-02209-y
This article is cited by
-
Small nuclear RNA genes in Mendelian disorders
Nature Genetics (2026)
-
De novo and inherited dominant variants in U4 and U6 snRNA genes cause retinitis pigmentosa
Nature Genetics (2026)
-
Clinical utility of genome sequencing in rare diseases: lessons from a single-center study of 1,452 Korean families
npj Genomic Medicine (2025)
-
Monoallelic and biallelic RNU4-2 variants in neurodevelopmental disorders
Journal of Human Genetics (2025)
