
Abstract
Copy number variable (CNV) genes are important in evolution and disease, yet their sequence variation remains a blind spot in large-scale studies. We present ctyper, a method that leverages pangenomes to produce allele-specific copy numbers with locally phased variants from next-generation sequencing samples. Benchmarking on 3,351 CNV genes and 273 challenging medically relevant (CMR) genes, ctyper captures 96.5% of phased variants with ≥99.1% correctness of copy number in CNV genes and 94.8% of phased variants in CMR genes. Ctyper takes 1.5 h to genotype a genome on one CPU. The ctyper genotypes give a 4.81-fold improvement in predictions of gene expression compared to known expression quantitative trait locus (eQTL) variants. Allele-specific expression quantified divergent expression in 7.94% of paralogs and tissue-specific biases in 4.68%. We found reduced expression of SMN2 due to SMN1 conversion, potentially affecting spinal muscular atrophy, and increased expression of translocated duplications of AMY2B. Overall, ctyper enables biobank-scale genotyping of CNV and CMR genes.
Similar content being viewed by others


Main
Human genomes frequently mutate through duplication and deletion, leading to copy number variation. Roughly 10% of protein-coding genes have shown copy number variation, with distinct distributions across populations1 and association with traits such as body mass index2 and disease, including cancer3, cardiovascular diseases4 and neurodevelopmental disorders5. While CNVs are infrequent genome wide, regions of long, low-copy repeats called segmental duplications are enriched in genes and are catalysts for recurrent CNVs due to non-allelic homologous recombination6,7. These regions include TBC1D3, NPIPA1 (NPIP) and NBPF1 (NBPF)8,9 and are associated with brain function and adaptation10,11. These regions additionally experience more rapid nucleotide substitutions than nonrepetitive DNA12. Consequently, studies only considering the aggregate copy number (aggreCN) of duplicated genes miss nonreference gene duplicates and variation among multiallelic CNVs, which have been shown to influence phenotypes and disease susceptibility13,14, including hypertension and type 2 diabetes15.
There are limited studies on variation between gene duplicates, particularly in studies using next generation sequencing (NGS). Existing CNV-calling tools rely on excess coverage, hiding variants among copies16. Furthermore, NGS alignments contain ambiguity and bias17 and miss divergent nonreference duplicates. New, high-quality pangenome assemblies provide sequence-resolved CNVs, including nonreference duplications, from diverse populations18,19,20. While graph-based pangenomes reduce reference bias21, they can merge similar sequences, including alternative alleles and functionally distinct paralogs, into shared sequences, obscuring paralog-specific variants and sequence diversity22. As pangenomes grow, alternative sequences, gene conversions and rearrangements will become more challenging to represent in graphs12, motivating the need for new tools and representations to analyze duplicated genes using NGS and pangenomes.
Here we present ctyper, a method that compares NGS reads to pangenome haplotypes to identify the most similar genomic segments between an NGS sample and haplotype assemblies and assign copy number state. The unmerged haplotype sequences preserve locally phased variants and capture complex variation, including structural variation and gene conversion. Here, we focus on complex, duplicated genes that are challenging to analyze using NGS and a reference genome. Leveraging alignment-free techniques and a polynomial time genotyping model, ctyper achieves both high accuracy and computational efficiency sufficient for future biobank analyses.
Results
Overview of the genotyping method
We represent variation as haplotype segments that are short enough to minimize disruption by recombination, allowing precise sharing with an NGS sample through identity by descent23 while capturing structural information including phased small variation, structural variation and gene conversion events. We label these haplotype segments as pangenome-derived alleles (PAs) and detect PAs shared with an NGS sample. PA boundaries are arranged to study variation of protein-coding genes. Each PA includes consecutive exons separated by <20 kb and 5 kb of flanking sequences (Methods), reflecting functional proximity of short-range transcription factors24 and population-level genomic linkage. PAs typically range between 10 and 100 kb, corresponding to the scale of linkage disequilibrium (LD) blocks25. While PAs generally correspond to individual genes, they also cover fractions of genes with long introns or, conversely, include tandemly arrayed paralogs within 20 kb.
For computational efficiency and to avoid alignment ambiguity in repetitive DNA, we use an alignment-free comparison of low-copy k-mers (DNA fragments of a fixed length k; k = 31) measured in NGS samples to genotype PAs. For each gene, we group all similar PAs in the pangenome, including orthologs, paralogs and homologous pseudogenes, and construct a matrix used in genotyping that contains the k-mer composition of all grouped PAs (Methods). The rows of a matrix correspond to individual PAs, columns correspond to k-mers exclusive to the grouped PAs, and cell values represent the k-mer multiplicity in each PA (Methods and Fig. 1a,b).

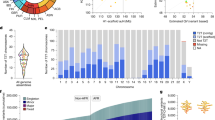
a, Demography of the reference pangenome assemblies. Map credit: Hogweard/Wikimedia Commons. b, Construction of pangenome k-mer matrices for CNV genes. Each individual gene is represented as a vector of counts of k-mers exclusively found among homologous sequences. All similar sequences including paralogs and orthologs are included and integrated as a k-mer matrix. c, Construction of phylogenetic trees based on k-mer matrices. d, Schematic of approach to estimate genotypes of alleles using NGS data. The k-mers from each matrix are counted in NGS data and normalized by sequencing depth. The normalized k-mer counts are projected to all pangenome genes. e, Reprojection of the raw results in the last step to integer solutions recursively based on the phylogenetic tree. f, An illustrative annotation and genotyping results on the SMN1 and SMN2 genes using HPRC samples. On the left side of the classification, the phylogenetic tree and heatmap of pairwise similarities are shown along with a mutation plot based on an MSA highlighting point differences in SMN1 in CHM13. All SMN genes are categorized into five major types and 17 subgroups. SMN1 and SMN2 correspond to the most common types of each paralog; SMN1-2, SMN1 partially converted to SMN2; SMN-conv, converted SMN genes, predominantly mapped to the SMN2 locus and found to be enriched in African populations; SMN2-2, a rare outgroup of SMN2. The GRCh38 assembly includes SMN1-2 and SMN2. The Phe280 T variant that disrupts the splicing of SMN2 transcripts is highlighted in red. The genotyping results of 1kGP continental populations are shown on the right. Rows correspond to subgroups, columns correspond to continental populations, and the colors of pie charts give distributions of copy numbers (CNs) among each continental population. AFR, African; AMR, American; EAS, East Asian; EUR, European; SAS, South Asian; ref, reference.
The genotyping is performed per matrix by identifying a combination of PAs (rows) and their copy number with the least-squared distance between their k-mer counts and that from an NGS sample. The sample k-mer counts are projected into the vector space of each k-mer matrix and assigned integer copy numbers using recursive rounding based on the phylogeny of PA sequences (Methods and Fig. 1c–e), resulting in a list of PA-specific copy numbers (paCNs). For example, there are 178 PAs for SMN genes, the gene family associated with spinal muscular atrophy. This includes copies of SMN1, SMN2 and paralogs with gene conversion26, for example, paralogs mapped to SMN2 that contain the SMN1 version of Phe280, the single nucleotide polymorphism (SNP) responsible for dysfunctional exon 7 splicing of SMN2 (ref. 27) (Fig. 1f).
PA database construction
We constructed a PA database for 3,351 genes previously reported as CNVs18,20 (Supplementary Table 1), using 114 diploid PacBio HiFi assemblies from the Human Pangenome Reference Consortium (HPRC), the Human Genome Structural Variation Consortium (HGSVC), the Chinese Pangenome Consortium (CPC) and two telomere-to-telomere assemblies28,29, in addition to GRCh38 and CHM13 (ref. 30). In total, we defined 1,408,209 PAs, organized into 3,307 matrices (Fig. 2a–c).

a, An example of amylase 1 PAs. Left: the corresponding order of all AMY1 PAs on assemblies, which are colored based on their major groups (unions of subgroups devoid of SVs >300 bp between members). Right: AMY1 genes are extracted as PAs as well as their flanking genes and sequences, including AMY2B translocated proximally to AMY1 and two pseudogenes: AMYP1 and RP5-1108M17. All PAs are vertically ordered according to the phylogenetic tree and aligned via graphic MSAs (Supplementary Notes). Homologous sequences are vertically aligned. Mutations are visualized as dots, and large gaps (deletions) are shown as spaces. Seven major groups are categorized including five paralogs and two orthologs. There are no pseudogenes around AMY1C, while AMY1A has RP5-1108M17 nearby and AMY1B has AMYP1 nearby. There are alternative versions of AMY1B and AMY1C with sequence substitutions. A new paralog called AMY1(Dup) is found primarily on haplotypes with duplications and has both pseudogenes nearby. Another paralog of AMY1 found with translocated AMY2B is called AMY1 + AMY2B. There are also two rare paralogs (blue and violet) and one singleton ortholog (steel blue). Alt, alternative; dup, Duplication. b, The size distribution of PAs on a log density. c, Circos plot of all PAs. Outer ring, the density of PAs in each Mb on GRCh38. Arcs, interchromosomal PAs are included in the same group. d, Annotation of PAs according to orthology and variants with respect to GRCh38. e, Identifiability of highly similar subgroups by unique k-mers. The total number of subgroups (blue) and the number of subgroups that may be identified by paralog-specific k-mers (red) are shown for each matrix with a size of at least three. f, Distribution of logistic pairwise divergence of PAs depending on orthology and phylogenetic relationship. The values shown are average values from each matrix. Small neighbor distances are an indicator of the strong representativeness of the current cohort. g, Saturation analysis for all subgroups using a recapture mode according to two sorted orders: African genomes considered first and non-African genomes considered first. The former order has a smoother curve than the latter, indicating that there are more African-specific subgroups.
Because of limited human genetic diversity and stronger LD across short distances, PAs are often highly similar or identical. To reduce dimensionality and facilitate cohort analysis, we used their phylogenetic relationships to merge similar PAs into highly similar subgroups (subgroups) treated as equal states (Methods). In total, we defined 89,236 subgroups, which were used to enumerate all PAs, analogous to human leukocyte antigen (HLA) nomenclature (Supplementary Fig. 1).
To annotate low-frequency variants and reference genome locations for orthologous or paralogous relationships, we mapped PAs to GRCh38 (Supplementary Notes). In total, 164,237 paralogous PAs across 6,389 loci were determined. Paralogous PAs that were similar to their corresponding reference locus (≥80% k-mer similarity) were labeled duplicative, and the remaining lower-identity paralogous PAs were labeled diverged. In total, 10,792 diverged paralogs from 2,734 subgroups were identified across 333 matrices (Fig. 2d). The divergent paralogs represent new sequences recalcitrant to canonical reference analysis. For example, some amylase PAs include paralogs for both AMY1 and AMY2B, reflecting an AMY2B translocation (Fig. 2a).
While most duplications were distal to their original genes, 6,673 PAs reflected proximal (<20 kb) duplications, including 1,646 PAs across 36 genes exhibiting ‘runaway duplication’ (ref. 31) with at least three proximal duplications (Supplementary Fig. 2). Proximally duplicated genes were included in the same PA as their ortholog as a heritable unit. Orthologous PAs were classified as reference alleles if they belonged to the same subgroup as the reference gene and as alternative alleles otherwise (Fig. 2d). All PAs were genotyped regardless of paralog–ortholog annotation so that the resulting genotypes contain population and copy number variation.
Ctyper databases capture population diversity
We assessed whether PAs capture unique aspects of genomic information that cannot be replicated by other CNV representations, including copy numbers of reference genes1,31, singly unique nucleotide k-mers1 (SUNKs) and large haplotype structures32,33. We found that PAs provide higher resolution of variation (for example, single-nucleotide variants), as 94.7% of variants are not reflected by sequences in GRCh38. Additionally, both nearby SUNK markers (Fig. 2e) and large haplotype structures were found to be poor proxies for PAs, and only a small proportion of PAs were found to link to SUNKs or larger haplotypes (Methods). Despite largely reduced dimensions, subgroups capture more than 80% of the total population variation (Methods and Fig. 2f). Finally, using saturation analysis34, we estimate that the current cohort represents 98.7% of subgroups in non-Africans and 94.9% in Africans (Methods), suggesting a near-saturated database (Fig. 2g).
Benchmarking genotypes from NGS samples
We genotyped 2,504 unrelated individuals and 641 offspring from the 1000 Genomes Project (1kGP). Most subgroups (99.25%) showed Hardy–Weinberg equilibrium (Methods and Fig. 3a) and thus little bias. There were 27 matrices with >15% subgroups in disequilibrium, which were mostly short genes (median = 4,564 bp) with few low-copy k-mers (Supplementary Table 2). Genotypes were accurate with an average F1 score for trio concordance of 97.58% (Supplementary Notes, Supplementary Table 3 and Fig. 3b), while 18 matrices had high discordance (>15%), primarily for subtelomeric genes or on sex chromosomes with poorer assembly qualities (Supplementary Table 4).

a, Hardy–Weinberg equilibrium of genotyping results from 2,504 1kGP unrelated samples. b, Genotype concordance of genotyping results from 641 1kGP trios, ordered by F1 error. The matrices with an F1 error of more than 15% were labeled by genomic location. c, Copy number comparison between assemblies and genotyping results from 39 HPRC samples shared with 1kGP. d, Sequence differences between genotyped and original alleles during the leave-one-out test using pairwise alignment of nonrepetitive sequences. e, Detailed leave-one-out comparison in the diploid telomere-to-telomere genome CN1. The results are categorized regarding the number of paralogs in CHM13 to show performances on different levels of genome complexity and the main sources of errors. The CN1 NGS sample had about 40-fold coverage. f, Ctyper runtime on all loci in CN1 for varying coverage. g, Benchmarking of HLA genotyping using ctyper on full and leave-one-out (LOO) databases, compared with T1K on 31 HLA genes. h, Benchmarking of CYP2D annotation on all CYP2D genes and CYP2D6 exclusively. FN, false negative; FP, false positive; TP, true positive.
We assessed copy number accuracy and bias among highly duplicated gene families (for example, amylase, NBPF, GOLGA and TBC1D3). The copy numbers derived from genotyping were compared to those from corresponding assemblies for 39 HPRC samples shared with the 1kGP using a database inclusive of these samples. To limit compounded error from misassembled sequences, we excluded samples with low-confidence sequences (Methods). For each sample, we benchmarked on all matrices for which the corresponding assembly was high in copy number (>10). The copy numbers were highly correlated (ρ = 0.996, Pearson correlation) with little bias (Fig. 3c), 0.2% missing copies (false negatives) and 2.4% additional copies (false positives), likely from unassembled genes in assemblies. High concordances remained when tests were expanded to all genotyped genes (ρ >= 0.996, Pearson correlation).
We assessed the sequence similarity of the genotyped alleles to the ground truth genome assembly for the 39 HPRC benchmarking genomes. Each sample was genotyped with the full database (full-set) or the database excluding its corresponding PAs (leave-one-out). We matched the genotyped PAs to the corresponding assembly PAs (Methods), excluding introns and decoys and sequences with <1 kb of nonrepetitive bases, and measured the similarity between the genotyped allele and the assigned query. We performed a similar analysis, treating the closest neighbor to each assembly PA from the database as the correctly genotyped locus. Due to mismatching from database sampling or misassemblies, 2.9% of PAs from the leave-one-out experiment and 1.0% from the full-set experiment were not paired with truth copies for assessment. For the full set, paired PAs had 0.36 mismatches per 10 kb, with 93.0% having no mismatches in nonrepetitive regions. The leave-one-out tests had 2.7 mismatches per 10 kb in nonrepetitive regions, which was 1.2 additional mismatches per 10 kb from the optimal solutions (closest neighbors); 57.3% of alleles had no mismatches, and 77.0% were mapped to the optimal solution (Fig. 3d). The leave-one-out results were 96.5% more similar to the original PAs than the closest GRCh38 gene at 79.3 mismatches per 10 kb.
To isolate sources of errors in cases of misassemblies, we directly compared leave-one-out genotyping results to a telomere-to-telomere assembly28 of genic PAs. The sample genotypes had 11,627 correctly matched subgroups, 599 (4.8%) mistyped to other subgroups, 131 from subgroups unique to the assembly (1.1%; out of reference), 127 false positives (0.5% F1) and 93 false negatives (0.4% F1) for a total F1 error of 6.7% (Methods and Fig. 3e), with a copy number agreement of 99.1%. This is a 3% increase in mistypes compared to trio discordance.
The computational requirements are sufficient for biobank analysis. The average runtime for genotyping 3,351 genes at 30× coverage was 80.2 min (1.0 min per 1× coverage for sample preprocessing and 0.9 s per gene for genotyping) on a single core (Fig. 3f) using ~20 GB of RAM, with support for parallel processing.
We compared the HLA, KIR and CYP2D6 genotypes to the locus-specific methods T1K35 and Aldy36. For 31 HLA genes, ctyper had an F1 score of 98.9% across all four fields of HLA nomenclature37,38 against the full-set analysis and a score of 86.3% for the leave-one-out analysis, while T1K had 70.8%. For protein-coding products (first two fields), ctyper reached 99.98% against the full-set analysis (with 99.9% copy number F1 correctness) and 96.5% (with 99.5% copy number F1 correctness) for the leave-one-out analysis, and T1K had 97.2% (Fig. 3g and Supplementary Tables 5 and 6). For 14 KIR genes, ctyper reached 98.5% across all fields in the full-set analysis and 70.6% for the leave-one-out analysis, while T1K had 32.0% due to the limited database. For protein-coding products (first three digits), ctyper reached 99.2% against the full-set analysis (with 99.9% copy number F1 correctness) and 88.8% for the leave-one-out analysis (with 99.2% copy number F1 correctness), while T1K had 79.6% (Supplementary Fig. 3). Benchmarking CYP2D6 star annotations of assemblies39, ctyper reached 100.0% against the full-set analysis and 83.2% for the leave-one-out analysis, compared to 80.0% using Aldy (Fig. 3h). There was perfect agreement of SNP variants for ctyper against the full-set analysis and 95.7% for the leave-one-out analysis, compared to 85.2% using Aldy.
Finally, we used ctyper to genotype 273 CMR genes40. Unrepetitive regions averaged 0.29 mismatches per 10 kb against the full-set analysis, 99.7% fewer than when comparing assemblies to corresponding GRCh38 sequences (baseline). The genotypes using leave-one-out databases had 4.9 mismatches per 10 kb, 94.8% fewer than baseline (Supplementary Figs. 4–6). Including repeat-masked low-complexity sequences (for example, variable-number tandem repeats), there were 10.5 mismatches per 10 kb against the full-set analysis (97.6% fewer than baseline) and 74.7 mismatches per 10 kb for the leave-one-out analysis (82.7% fewer than baseline; Supplementary Figs. 7–9).
We compared genotyping of HLA and CMR genes to a contemporary method using pangenomes, Locityper41, using leave-one-out analysis. For HLA, Locityper achieved an F1 score of 87.9% (versus ctyper, 86.3%) for predicting all four nomenclature fields, while ctyper performed slightly better on the first two fields for protein-coding variants (96.5% versus 94.0%; Supplementary Notes), although ctyper had a roughly 218× speedup due to alignment-free genotyping. When analyzing CMR genotypes, there were 19.8 fewer mismatches per 10 kb than the Locityper genotypes in comparable regions (Supplementary Notes, Supplementary Table 7 and Supplementary Fig. 10).
Sequence-level diversity of CNVs in global populations
We used principal-component (PC) analysis (PCA) to examine the population structure of PA genotypes in the 2,504 unrelated 1kGP samples, 879 Genotype–Tissue Expression (GTEx) samples and 105 diploid assemblies (excluding HGSVC due to quality filtering), excluding rare subgroups (<0.05 allele frequency) and limiting copy number to ten to balance the weights of PCs (Fig. 4a). All data cluster by population as opposed to source, suggesting little bias between genotyping and assembly or across NGS cohorts. The top 0.1% highest-weighted subgroups in PC1 have an average aggreCN variance of 26.33, significantly larger than the overall of 4.00 (P value = 1.11 × 10−16, F-test). Similarly, PC2 and PC3 have mean aggreCN variances of 19.73 and 7.20, suggesting that CNVs are weakly associated with sequence variants. Furthermore, PC1 is the only PC that clustered all samples into the same sign with a geographic center away from 0, suggesting that it corresponds to modulus variance (hence aggreCN) if treating samples as vectors of paCNs. Meanwhile, PC2 and PC3 were similar to the PCA plots based on SNP data of global samples42, suggesting that they are associated with the sequence diversity of CNV genes. The total number of duplications is elevated in African populations (Fig. 4b), reflected in the order of PC1 (Fig. 4a).

a, PCAs of allele-specific copy numbers on the union of PA genotyping results and assembly annotations. b, Distribution of total autosomal gene copy numbers among unrelated 1kGP samples, including pseudogenes (AFR, n = 685; admixed AMR, n = 352; EAS, n = 511; EUR, n = 522; SAS, n = 516; box shows median and interquartile range; whiskers extend to 1.5× interquartile range, with outliers beyond). c, Population differentiation measured by F statistics of duplications among different continental populations. Genes with a paralogous subgroup with an F statistic of more than 0.35 are labeled. d, Mean absolute variation in copy numbers and RPD in sequences. Based on our genotyping results from unrelated 1kGP genomes, for genes found to be CNV to the population median in more than 20 samples, we determined the average aggreCN difference (MAE) between individuals and estimated the average paralog difference in sequences relative to the ortholog difference. e, mLD between pairs of CNV genes less than 100 kb apart. The larger MAE value of each pair is used for the x-axis values. The total locus length denotes the length from the beginning of the first gene to the end of the last gene.
We examined ctyper genotypes to measure the extent to which duplications show population specificity. We used the F statistic, a generalization of FST that accommodates more than two genotypes (Methods), to test the differences in distributions across continental populations (Fig. 4c). In total, 4.4% (223 of 5,065) of duplicated subgroups showed population specificity (F statistic > 0.2; Supplementary Table 8). The subgroups of PAs with the highest F statistic (0.48) contain duplications of HERC2P9, a known differentiated gene7. Additionally, a converted copy of SMN2 annotated as a duplication of SMN1 is enriched in African populations (F statistic = 0.43).
We then measured the divergence of duplicated genes from their reference copies, indicating recent or ancient duplications and providing a measure of reference bias from missing paralogs. We constructed multiple-sequence alignments (MSAs; Methods) for sequences of each matrix and measured all pairwise differences in nonrepetitive sequences. We determined the average paralog sequence divergence relative to the ortholog divergence (Methods), which we refer to as the relative paralog divergence (RPD). We also measured copy number diversity using mean absolute error (MAE), indicating the CNV level among populations (Fig. 4d). Based on RPD, using density-based spatial clustering of applications with noise43, we identified two peaks at 0.71 and 3.2, with MAE centers at 0.18 and 0.93, corresponding to genes with rare and recent CNVs and more divergent and common CNVs, respectively. The latter reflect CNVs on different structural haplotypes that cannot be analyzed using a single reference genome. For example, AMY1A has a high RPD at 3.10 because of truncated duplications. These results are consistent with ancient bursts of duplications in human evolution44.
We next used ctyper genotypes to investigate recombination at different CNV loci. We determined multiallelic LD45 (mLD; Methods) between PAs using the unrelated 1kGP samples for 989 subgroups that were adjacent less than 100 kb apart in GRCh38 and reported the average mLD within each matrix (Fig. 4e). There was a stronger negative rank correlation between MAE of copy number and mLD (ρ = −0.24, P value = 3.4 × 10−15, Spearman’s rank) than the rank correlation between mLDs and locus length (ρ = −0.21, P value = 1.5 × 10−11, Spearman’s rank), suggesting a reduced haplotype linkage in genes with frequent CNVs. The lowest mLD (0.013) was found in FAM90, a gene with frequent duplications and rearrangements46. The 29 loci with highest mLD (mLD > 0.7) were enriched in the sex chromosomes (n = 19). Furthermore, HLA-B and HLA-DRB had mLD > 0.7 and only deletion CNV (Supplementary Notes).
eQTL analysis
To investigate the impact of paCNs on expression, we performed eQTL analysis using the Genetic European Variation in Disease47 (GEUVADIS) and GTEx48 cohorts. There were 4,512 genes that could be uniquely mapped in RNA-seq alignments. An additional 44 genes, such as SMN1, SMN2, AMY1A, AMY1B and AMY1C, have indistinguishable transcription products and were analyzed by pooling among all copies. We assigned PAs to these transcripts based on exonic sequences and performed association analysis with paCNs (Methods and Supplementary Table 9).
After merging paCNs to aggreCNs, 5.5% (178 of 3,224) of transcripts showed significance (corrected P = 1.6 × 10−5, Pearson correlation) as previously observed31. By contrast, when updating aggeCNs by individual paCNs and performing multivariable linear regression on expression (Methods), there were significant improvements in fit for 27.6% (890 of 3,224) of transcripts (corrected P = 1.6 × 10−5, one-tailed F-test; Fig. 5a). To test whether the fit was explained by the nonuniform expression of different alleles of the same reference gene, we used a linear mixed model (LMM; Methods) to regress total expression to individual subgroups and estimate allele-specific expression and then compared these values to other subgroups of the same matrix that were assigned to the same reference gene (Supplementary Table 10). For subgroups within solvable matrices and more than ten samples, we found that 7.94% (150 of 1,890) of paralogs and 3.28% (546 of 16,628) of orthologs had significantly different expression levels (corrected with sample size = number of paralogs + orthologs, corrected P = 2.7 × 10−6, χ2 test; Fig. 5b). Overall, paralogs were found to have reduced expression (Fig. 5c), consistent with previous findings for duplicated genes49.

a, Q–Q plot of associations of (blue) aggreCNs with gene expression in GEUVADIS samples (red) and the improvement of allele-specific copy number over aggreCN. b, Comparative gene expression Q–Q plots of orthologs (blue) and paralogs (red). c, Fold change effect size of all significant alternative expressions in b. Fold changes as well as P values are shown. Top: density of fold change effect size of orthologs and paralogs. d, Preferential tissue expression of orthologs and paralogs. e, Top: comparison of different models for explained expression variance (R2). Bottom: quantification of variance explained by different representations at different levels of CNV frequencies: full paCN genotypes, aggreCN and known eQTL variants (var.). f, Case study on SMN genes showing decreased gene expression in SMN-converted. The average expression level in PEER-corrected GEUVADIS samples (n = 386) is shown under different copy numbers of SMN1 (n = 741), SMN2 (n = 569) and SMN-converted (n = 89). Transcript levels are the total coverage of all isoforms, and the exon 7 splicing level is measured by counting isoforms with a valid exon 7 splicing junction. g, Case study on amylase genes showing increased gene expression of translocated AMY2B using PEER-corrected GTEx pancreas data (no duplications, n = 209; ordinary duplications, n = 6; AMY2B to AMY1, n = 25; AMY2B to AMY2A, n = 4; RNA-seq samples, n = 304; box shows median and interquartile range; whiskers extend to 1.5× interquartile range, with outliers beyond).
We compared expression in 57 tissues in the GTEx samples to test for preferential expression of paralogs (Methods and Supplementary Table 11). There was alternative tissue specificity for 132 of 2,820 paralogs (4.68%) and 225 of 19,197 orthologs (1.17%) (corrected P = 6.4 × 10−8, union of two χ2 tests; Methods and Fig. 5d).
Additionally, we used analysis of variance (ANOVA) to estimate the proportion of expression variance (R2) explained by paCNs in GEUVADIS expression data and compared it to that in a model based on known SNPs, indels and eQTL structural variants (SVs)50 (Methods). As expected, the highly granular paCNs explained the most variance: on average, 10.3% (14.3% including baseline). By contrast, 58.0% of transcripts are genes with known eQTL variants that explained valid variance by 2.14% (1.60% considering experimental noise, in agreement with a previous estimate of 1.97%51). On average, 1.98% of the variance was explained by aggreCNs, and 8.58% was explained by subgroup information. When combining both paCNs and known eQTL sites, 10.4% (19.0% including baseline) of the valid variance was explained (Fig. 5e).
We examined the SMN and AMY2B genes as case studies due to their importance in disease and evolution27,52. The SMN genes were classified as SMN1, SMN2 and SMN-converted. We found no significant difference between the expression of all transcripts of SMN1 and SMN2 (0.281 ± 0.008 versus 0.309 ± 0.009; P = 0.078, χ2 test). However, significant differences were found between SMN-converted, and SMN1 and SMN2 (0.226 ± 0.012 versus 0.294 ± 0.002; P = 1.75 × 10−7, χ2 test), with a 23.0% reduction in SMN-converted expression. By contrast, despite having lower overall expression, SMN-converted had 5.93× the expression of valid exon 7 splicing53 of SMN2 (P = 2.2 × 10−16, χ2 test), indicating that SMN-converted has full functional splicing54 but lower overall expression (Fig. 5f). We studied the expression of AMY2B duplications, including alleles translocated proximally to other AMY genes, such as the PAs containing AMY1 and AMY2B in Fig. 2a. Using probabilistic estimation of expression residuals (PEER)-corrected GTEx pancreas data, we found that translocated AMY2B genes had significantly higher expression than other duplications (1.384 ± 0.233 versus −0.275 ± 0.183, P = 7.87 × 10−9, χ2 test) (Fig. 5g).
Discussion
New pangenomes present both opportunities and challenges to study complex genetic variation (for example, CNVs, recurrent SVs, translocations and gene conversion): they reveal the landscape of complex variation but require new tools for representation and analysis. We represent genomic variation as PAs: haplotype segments that capture genomic structural information and phased variation. To support large NGS cohort analyses, we developed an alignment-free genotyping tool, ctyper, to genotype PAs with NGS, providing allele-specific sequence information and copy number. The genotyping is based on a new mathematical model that relaxes an nondeterministic polynomial time (NP)-hard problem into a more efficient polynomial semianalytic solution with robust genotypes and copy number estimates. Although the analysis here focuses on CNV genes, ctyper is applicable to genotype complex genetic variation and local phasing genome-wide.
The use of ctyper genotypes increases the scope of NGS studies to profile variation in unmappable CMR and CNV genes. For example, our finding that CNVs reflect two modes of variation (highly similar (recent) and low-identity (ancient and polymorphic) duplications) is based on the 1kGP genotypes rather than assembly annotation. Additionally, ctyper genotypes yield tissue-specific expression of paralogs and relative contributions to expression from different duplication structures, for example, SMN genes.
We investigated the significant improvement of ANOVA on PAs, the genotypes of which are multiallelic and reflect different combinations of variants in contrast to conventional known biallelic eQTL variants. First, compared to PAs, there were either very few or very many eQTL variants per gene, indicating LD (Supplementary Fig. 11) as addressed by fine-mapping55 and, in the latter case, increasing multiple-testing burden56. Indirect association due to LD also explains why there was a greater proportion of variance explained among genes with more CNVs by conventional eQTL variants (for example, the HPR genes; Supplementary Fig. 2). However, as the frequency of CNVs increases, the variance explained by eQTL variants increases (t = 3.80, P value = 1.6 × 10−4, Pearson’s correlation) and the number of eQTL variants decreases (t = −4.79, P value = 2.1 × 10−6, Pearson’s correlation), suggesting that larger effects such as CNVs might overshadow the discovery of other eQTL variants not in LD (the increase in total variance reduces significance in association analysis using a Gaussian-like model). Because PAs incorporate LD, they will suffer less from such LD-based problems. Furthermore, gene expression might not be a linear additive effect of variants57. For example, although SMN-converted contains variants from SMN1 and SMN2, its overall expression is lower than both. In this manner, using a genetic model with linked variants such as PAs improves upon the linear additive model in predicting gene expression. Because these limitations also apply to non-CNV genes, the concept of PAs may have a wider potential for future association analysis.
Due to the limited sample size, our associations are based on subgroups rather than individual PAs. Different cohort sizes may require different levels of subgroup granularity. Our current granularity was designed for biobank cohorts; studies in smaller cohorts may need to define broader subgroups. For example, the three subtypes of SMN-converted showed little difference in our eQTL analysis and were merged in our case study, but larger cohort studies may find their differences. The granularity of genotyping is additionally defined by the length of PAs; shorter PAs more accurately reflect small variants, while longer sequences preserve more structural information and may be preferable in regions with low recombination, such as HLA-DRB.
Methods
Constructing the pangenome-derived allele database
Search and extraction of initial genes of interest from pangenome assemblies
Our pangenome cohort was composed of assemblies from the HPRC (92 haplotypes, excluding HG02080 due to abundant flagged regions), the CPC (114 haplotypes), the HGSVC (18 haplotypes; only PacBio HiFi assemblies were used), two telomere-to-telomere diploid assemblies (four haplotypes) and reference genomes (GRCh38 including alternative loci and T2T-CHM13). The gene coordinates used were from GENCODE version 39 based on the GRCh38 reference genome.
We constructed databases for 3,203 genes found to have copy number variation in the HPRC and CPC studies. Genes were initially organized into ‘query sets’ where each query set encompassed genes with functional or similar sequences including pseudogenes and genes with distant homology within the same gene family. The query sets were initially defined based on genes with shared name prefixes and were used to locate all similar sequences within the pangenome.
Efficient mapping methods58,59 missed alignments to sequences that contain k-mer matches and decreased genotyping accuracy when not included in our database, including small pseudogenes and diverged paralogs. To address this, we developed a sensitive and efficient scanning scheme centered on k-mer clusters to detect all similar sequences for genes of interest in the pangenome (Supplementary Notes).
The hotspots defined by k-mers often include loci mapped by multiple genes from a query set and tandemly duplicated genes. To account for this redundancy, we merged alignments that were less than 10 kb apart (together with 5 kb of flanking sequences, this merges genes within 20 kb), causing tandemly duplicated genes to be merged into a single locus. To avoid genotyping longer loci that may be split by recombination, we divided loci at the midpoints of introns exceeding 20 kb. To ensure a minimum locus length, flanking sequences were adjusted to achieve a minimum length of 15 kb. These methods aim to standardize the size of each sequence to approximate the size of LD blocks. The collection of all sequences mapped by a query set are referred to as initial matrix sequences.
Filtering and polishing initial matrix sequences and k-mers
For each genome, we first extracted k-mers found exclusively in the initial matrix sequences. We then filtered out low-complexity k-mers with a composition of at least two-thirds redundant 2-mers or 3-mers and k-mers with high (>70%) or low (<30%) GC content60 to reduce bias in genotyping. The matrix sequences composed of a majority of filtered k-mers as well as those from HPRC nonconfident regions, truncated sequences from small scaffolds and non-telomeric sequences within 10 kb of the end of a scaffold were removed.
The initial groups of matrix sequences had sequences with low homology but similar names. Unrelated sequences from our initial groups of matrix sequences were partitioned using graph partitioning based on k-mers (Supplementary Notes).
The resulting filtered sequences were labeled as PAs. There were 1,408,209 PAs for 3,351 genes in total from 3,307 partitions. This includes any additional genes not defined as duplicated in the original set that had high sequence similarity. The average PA length was 33 ± 29 kb and included protein-coding genes (69%), processed pseudogenes (20%), intronic duplications (5%) and decoys (unrelated genes that share homology and improve genotyping accuracy when included; 7%). We represented each final partition as a single matrix along with the list of low-copy k-mers specific to the matrix that passed filtration (k-mer matrix). Each row corresponds to a PA sequence. Each column corresponds to a distinct k-mer. The matrix values are the counts of corresponding k-mers in the respective PA. The counts are mostly 0 or 1 but are occasionally greater when there are low-copy repeats in the PA or the row represents a tandemly duplicated locus.
Annotation of pangenome-derived alleles
k-mer-based phylogenetic tree construction
We constructed a separate phylogenetic tree for each k-mer matrix for use in annotation and genotyping. For computational efficiency as well as consistency with our k-mer-based genotyping and annotation, we used distances based on k-mers instead of MSAs for construction.
The matrix structure (we use M to denote any arbitrary k-mer matrix) allows us to easily measure the concordance between any two sequences by their vector form, Gi and Gj, by calculating their inner product, denoted as <Gi × Gj>. The norm matrix N = M × MT reflects the k-mer concordances for all sequence pairs within the matrix. We constructed a similarity matrix, S, where Si,j is the cosine similarity of Gi and Gj. Finally, we used the unweighted pair group method with arithmetic mean algorithm on S to generate the phylogenetic tree for each partition.
Clustering of pangenome-derived alleles into highly similar subgroups
For each group of sequences corresponding to a matrix, we used its corresponding phylogenetic tree for the annotation and classification of highly similar groups of alleles, which we term ‘highly similar subgroups’. The classification of highly similar subgroups is guided by two criteria: (1) a subgroup must have homology among the members. This is quantified by ensuring that the largest k-mer distance between any two members does not exceed 155 k-mers (roughly equivalent to the variation caused by five single-nucleotide variants) or a SV of approximately 95 bp; subgroups represent most common variants within ~30 kb. (2) Each subgroup must be distinct from neighboring subgroups. This is measured using a k-mer F statistic score, which must exceed 2 when compared with adjacent subgroups. In cases in which subgroups are composed of fewer than three members, the F statistic may not be reliable. We default this score to 0 for small subgroups but change the cutoff of the former criteria to 155 × 3 to detect singleton rare events.
We applied these criteria to all clades in a ‘bottom–up’ recursive approach starting from leaves to report the largest possible highly similar subgroups.
Pangenome-derived allele annotation relative to the reference genome
We annotated CNV events and duplicated alleles in the pangenome assemblies relative to the GRCh38 reference genome. This requires solving for orthology assignment61, a challenging task because PAs often align to multiple paralogs on GRCh38 and the orthologous gene identified by reference mapping may not be the most similar reference gene due to gene conversion or translocation (Figs. 1f and 2a). Here, we match PAs to their closest GRCh38 genes based on k-mer similarity. For every haplotype, we obtained pairwise similarities between the haplotype and GRCh38 PAs. We matched PAs to their most similar GRCh38 PAs, starting from the most similar pair, until all PAs were matched or failed to match (had no reference gene with >90% similarity). Matches to reference genes that had already been matched were annotated as duplications.
The PAs that formerly failed to match were likely alleles with large SVs. We attempted to map them back to GRCh38 using 100-kb flanking sequences and a two-step liftover. First, we lifted PAs to the region with the best local alignment coverage, allowing SVs in alignments. Next, we performed global pairwise alignment between PAs and the lifted region to locate the best-aligned gene considering local translocations and tandem duplications (Supplementary Notes).
Finally, to annotate the proximal duplications as well as to identify diverged paralogs that failed to match by both previous methods, we annotated PAs using gene transcripts to identify PAs containing genes, pseudogenes and putative protein-coding genes. We aligned all exons from the same matrix to PAs and, based on the exon order and alignment scores, determined the optimal combinations of transcripts for each PA (Supplementary Notes). PAs containing no exons were annotated as introns, and PAs containing only transcripts of other unrelated genes were annotated as decoys.
Classification of orthologs and paralogs in the pangenome
PAs were classified into four categories, including two types of orthologs and two types of paralogs for downstream analysis:
-
1.
Reference alleles: alleles in the same subgroup as GRCh38 alleles with almost identical sequence.
-
2.
Alternative alleles: orthologs at the same genomic locus as the reference gene but in different subgroups from GRCh38 alleles due to sequence divergence or structural variation, such as HPR, NBPF and CYP2D6.
-
3.
Duplicated paralogs (alleles): paralogs duplicated to different loci from their source genes but retaining high sequence similarity (>80% k-mer similarity), reflecting recent segmental duplications. For example, AMY1A, AMY1B and AMY1C are still often considered functionally the same despite their distinct locations.
-
4.
Diverged paralogs (alleles): paralogs duplicated to different loci from their source genes and significantly divergent (<80% in k-mers). These are characterized by highly diverse nonreference paralogs, incomplete gene duplications and new divergent processed pseudogenes. An example of diverged paralogs is at a translocation event between AMY1 and AMY2B.
Justification of the representation of pangenome-derived alleles and highly similar subgroups
Comparison of pangenome-derived alleles with other genomic representations
For each PA, we compared the nearest neighbor in our pangenome database as a proxy for the optimal genotyping result of samples containing that PA to its closest GRCh38 gene based on k-mer similarity. The nearest neighbor had 94.7% fewer differences on average compared to GRCh38 matches, and 57.3% had identical nearest neighbors.
There were 38.8% of subgroups with more than three members that were identifiable by k-mers uniquely shared in the subgroup, analogous to SUNKs (Fig. 2e). For example, no SUNKs exist between SMN1, SMN2 and SMN-converted due to gene conversion (Fig. 1f).
We found that recombination or other structural variation creates unique combinations of PAs that cannot be represented during leave-one-out analysis. For example, in the amylase gene, 40% (90 of 226) of haplotypes could not be represented with the remaining subgroups, particularly those with a greater number of copies than GRCh38 (45 of 67). When all PAs devoid of SVs were considered equally in a single large subgroup, 20% (46 of 226) of haplotypes remained singleton, especially those with additional copies (26 of 67). Furthermore, new subgroups are found within different structural haplotypes, such as the PAs containing adjacent AMY1 and AMY2B due to rearrangement (Fig. 2a).
Justification of highly similar subgroups in representing population diversity
We measured the extent that highly similar subgroups capture sufficient population diversity. The average pairwise k-mer cosine similarity was 98.8% within each highly similar subgroup (one base change adds ~k differences), compared with an average 94.2% cosine similarity to the corresponding reference sequence (a 5.03× decrease). Between two phylogenetically neighboring subgroups having at least three members each, the between-group variance was 6.03× greater than the within-group variance, showing that most genetic diversity may be represented using a small number of haplotype states, as both criteria suggest that more than 80% of total population variation could be represented by highly similar subgroups.
Genotyping NGS samples with ctyper
Initial solution based on linear regression
Given an NGS sample and a k-mer matrix M derived from PAs, we generate a vector V of corresponding k-mer counts from the NGS sample, normalized by sequencing coverage. We seek to find a vector X that denotes the copy numbers of all PAs and minimizes the squared distance to the k-mer counts observed in NGS data, for example, argminx (ǁMT × X − Vǁ). The integer solution through mixed-integer linear programming is NP hard6; however, the relaxed non-integer solution based on squared distances has an efficient analytic solution. Compared with absolute distance, squared distance is more suitable for the normal-like noise in NGS data62,63.
To make the solution closer to the maximum likelihood estimate, during the regression, we rescale the weights of k-mers to even out their expected uncertainty. Assuming that the observation of k-mer copy number follows a negative binomial distribution with the dispersion small enough to be distinct from Poisson63, the expected variance is roughly proportional to the square of observation; therefore, we weight k-mers by the square of the reciprocal of their observed copy number. We also apply smaller weights (adjust = 0.05) to k-mers observed in only one PA and not in NGS because they are more likely to be assembly errors.
Integer solution based on recursive phylogenetic rounding
Initial linear regression yields solutions in the form of small floating point values, in which the alleles with the highest coefficients are not necessarily those closest to query genes. However, as shown by mathematical analysis (Supplementary Notes), there are strong relationships between the initial least-error solution and the true integer solutions under a phylogenetic framework:
-
1.
Nonnegative solutions: without uncertainty in predicting k-mer copy numbers, the least-error solution should be nonnegative. Therefore, we obtain a nonnegative least-error solution via the Lawson–Hanson algorithm64.
-
2.
Total copy number estimation: the sum of the initial solutions should approximate the total number of the true integer solutions, allowing us to estimate the total gene copy number in the querying sample.
-
3.
Phylogenetic position prediction: on a binary phylogenetic tree, the branch with a shorter vector distance to the genes in the querying sample will have a larger sum of coefficients (inversely proportional to distance). This relationship enables us to predict the phylogenetic position of each gene in the query sample.
-
4.
Fractality of the least-squared error solution on a phylogenetic tree: if a solution is the least-squared error solution of the tree, it is also the least-squared error solution within each clade, allowing the greedy method to perform on the phylogenetic tree.
-
5.
Large database effect: in large databases, having more genes highly similar to query genes increases condition number and tends to distribute the total coefficients across them, resulting in smaller individual coefficients. However, the total sum of these coefficients increases, improving the precision of phylogenetic position prediction, and this effect does not plateau.
-
6.
With sequencing coverage variance for NGS at ~30-fold coverage, the model precision remains. Sequenced variance is not the primary source of error.
Given the high ‘convergence’ and fractality of the solution on the phylogenetic tree in large databases, we developed a greedy algorithm to efficiently convert non-integer solutions into integer solutions. This iterative algorithm follows a bottom–up approach, starting from the leaves and progressing toward the root. At each hierarchical level, non-integer values are rounded to the nearest integer solution that minimizes the overall residual, while any remainder is propagated to the next level. Because, at each level of the hierarchy, there are only two remainders from either branch of the tree, this solution is highly efficient. We label this approach as recursive phylogenetic rounding. The pseudocode for this algorithm (naive version and optimized version) is provided in the Supplementary Information.
Benchmarking of genotyping
Hardy–Weinberg equilibrium
Hardy–Weinberg equilibrium analysis was performed on autosomal chromosomes, setting the maximum copy number to two and testing for significance using the χ2 distribution.
Comparison of genotyping results to pangenome assemblies
The accuracy of PA genotypes was measured by aligning the genotyped PA sequences to the corresponding assembly (ground truth). The assembly PAs were paired with genotyped PAs by a greedy method (Supplementary Notes) and aligned using Stretcher65 for masked sequences and a pairwise alignment method distributed with Locityper41 for unmasked sequences. This determined the number of mismatched bases in unmasked regions that correspond to ctyper k-mer queries.
Classification of errors
We classified four types of errors for our benchmarking:
-
1.
False positive: the genotyping results have an additional copy;
-
2.
False negative: the genotyping results have a missing copy;
-
3.
Mistyping: copy assigned to an incorrect type;
-
4.
Out of reference: the sample has a PA that is a singleton subgroup and excluded from the genotyping database during leave-one-out analysis.
Benchmarking HLA, KIR and CYP2D genes with public nomenclatures
We labeled all IPD-IMGT and CYP2D-star annotations for PAs. HLA and KIR genes in assemblies were annotated using Immuannot66, and CYP2D6 was annotated using Pangu39. We annotated genotyped PA sequences and compared them to the assembly annotation from matched samples. The results for HLA were compared with T1K35, and the results for CYP2D6 were compared with Aldy36 (Supplementary Notes).
Population analysis of pangenome-derived alleles
Total number of duplication events from genotyping results
We calculated the total number of duplication events for each 1kGP unrelated sample from ctyper genotypes, excluding seven samples that had a population mean more than five standard deviations above the average. The total number of each reference gene including pseudogenes and pseudogene-like exonic fragments was counted in each genome and compared to that in GRCh38, excluding alternate haplotypes. Each duplication event was called if the genome had a copy number more than twice that of GRCh38, excluding decoys, introns and sex chromosome genes. The total number of duplication events was reported for each genome.
Measuring F statistic values
We used the F statistic to measure the population specificity of subgroups. The F statistic is based on the F-test, with which we obtained the variances of copy numbers within all continental populations (within-group variance) and used them to divide the variances of copy numbers across different populations (between-group variance).
Relative paralog divergence
RPD measures the mean divergences of the paralogs to other alleles, in relation to the mean divergence between only orthologs. RPD was determined for each reference gene and based on the graphic MSAs (Supplementary Notes) of PAs assigned to that reference gene as well as the ctyper genotyping results.
The divergence was first determined for each pair of PAs assigned to the same reference gene based on the alignment scores of unmasked bases (mismatch and gap open = −4 and gap extend = 0, normalized by total alignment length) from graphic MSAs. The mean divergence of orthologs was determined by averaging divergence values between the two PAs from samples with copy number = 2. Samples were divided into those with additional copy numbers (copy numbers more than the population median for a gene) and those with no additional copy numbers otherwise.
It is challenging to distinguish paralogs from orthologs in complex rearrangements (for example, Fig. 2a). To only obtain divergence values from additional copies, we performed statistical estimation based on large populations. The mean sequence divergence from samples with no additional copy numbers was used as a baseline B. When the population median copy number = Y, because there are Y(Y − 1)/2 pairs, then the total baseline is B × Y(Y − 1)/2, which is subtracted from total divergence values of samples with duplications, and Y(Y − 1)/2 is subtracted from the total number of pairs. For a sample with copy number = X, the estimated paralog divergence is (total variance − B × Y(Y − 1)/2)/(X(X − 1)/2 − Y(Y − 1)/2).
The mean paralog divergence value was determined for all samples with additional copy numbers and normalized by the mean divergence of the orthologs.
Multiallelic linkage disequilibrium
mLD is an analytic continuation of SNP-based biallelic LD to allow computing linkages between multiple genotypes on neighboring loci. We measured LD between each pair of genotypes across both loci and took the weighted average of all pairs as the product of both allele frequencies of pairs.
Expression analysis of pangenome-derived alleles
Determining transcripts for expression analysis
We first represented each gene by its major transcripts from the Matched Annotation from NCBI and EMBL-EBI67 project and then aligned individual exons. Transcripts were recursively clustered together if they overlapped with previously clustered transcripts with more than 98% overall similarity, taking the average similarity of all aligned exons from the transcripts. We considered these clusters as the same transcript even though they were from different genes. Third, for each transcript, we identified all its exons and searched for unique exons that did not overlap with exons from other transcripts. Fourth, we used these unique exons to represent each transcript and filtered out transcripts that had no unique exons (2,079 of 2,579 filtered genes were known pseudogenes). Lastly, we assigned PAs to each transcript if they contained any of the corresponding unique exons with at least 98% similarity.
Expression correction
Following precedent68, we logistically corrected the raw transcript-per-million GTEx values using the tool PEER together with the first three PCs from GTEx. For GEUVADIS samples, we obtained PCs from PLINK version 2 (ref. 69) with default settings on genotypes in chr1 (ref. 70). For cross-tissue analysis, we corrected raw transcript-per-million values using DESeq2 (ref. 71) with default settings.
Association between CNVs with gene expression
We first associated gene aggreCN with expression using Pearson correlation (linear fit). To test whether including allele-specific information improved fit, we replaced aggreCN with the ctyper paCNs to perform multivariable linear regression using paCNs as dependent variables and gene expression levels as independent variables. We compared the residuals of multivariable linear regression with residuals from the former linear fitting (F-test), reporting the one-tailed P values of the reduced residual corrected for the number of transcripts tested (n = 3,224).
Linear mixed model
We performed LMM to estimate the individual expression of each subgroup with y ≈ Xβ, where y is the total gene expression, X is copy numbers and β is subgroups, solved using ordinary least-square regression.
Alternative expression of subgroups
Paralogs were assigned to a GRCh38 reference gene based on exon annotation. We merged all other subgroups assigned to the same GRCh38 gene into a single variable separate from the subgroup then being tested. Additionally, we included paralogs assigned to other reference genes that might also influence total expression to adjust for interference. For subgroups within solvable matrices with more than ten nonzero expression values, we regressed the expression values to all variables to measure effect sizes using the R lm function72. We then compared the effect sizes between the currently tested subgroup and the other subgroups of the same gene (χ2, linearHypothesis package73), corrected for the number of total subgroups tested (n = 18,518).
Across-tissue expression comparison
We determined whether a subgroup had an alternative most-expressed tissue compared to other subgroups of the same gene, using previously gene assignment and expression filtering to calculate alternative expression of subgroups. We estimated the gene expression level of each subgroup within each of the 57 tissues in GTEx version 8 using LMM analysis. The tissue with the highest expression was compared to the tissue with the second highest expression (χ2). We then compared the results between the currently tested subgroup and all other subgroups of the same gene to see whether they had a different highest-expressed tissue. When the highest-expressed tissues were different, we tested the P value of either event by combining the P values from both sides as Pcombined = P1 + P2 − P1 × P2 and corrected for the number of tests on all tissues (n = 776,902).
Analysis-of-variance tests on gene expression
We first measured the total expression variance for each eQTL transcript in the GEUVADIS cohort, filtering out units with per-sample variance less than 0.1. Experimental noise was estimated by measuring expression variance between different trials of the same individuals (mean = 10.5% of the total variance) and excluding transcripts with experimental noise exceeding 70% of the total variance, resulting in 639 transcripts. We applied the one-in-ten rule to restrict the number of variants tested to be not greater than 45 (10% of the sample size) to avoid overfitting, filtering 18 transcripts. When there were more than 45 known eQTL variants, we used the 45 variants with the lowest P values. The valid expression variance was obtained by subtracting experimental noise from the total expression variance. Using ANOVA, we estimated the explained valid variance and adjusted the results by subtracting a baseline, defined as the mean expression variance explained by permuting the orders of all samples (estimated by the mean of 100 trials). If there were no reported eQTL variants, a value of 0 was used for known eQTL variants.
The variance explained by gene aggreCN was measured by subtracting the average of 100 ANOVA results on randomly permuted subgroups from the total explained variance by paCNs to obtain the variance explained by subgroup information.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Allele database and annotations can be found on Zenodo (https://doi.org/10.5281/zenodo.13381930)74.
Code availability
Software can be found on GitHub (https://github.com/ChaissonLab/Ctyper). Benchmarking and analysis code can be found on GitHub (https://github.com/Walfred-MA/CNVAnalyze). Software and analysis can be found on Zenodo (https://doi.org/10.5281/zenodo.16956833)75.
Change history
16 February 2026
A Correction to this paper has been published: https://doi.org/10.1038/s41588-026-02518-w
References
Sudmant, P. H. et al. Diversity of human copy number variation and multicopy genes. Science 330, 641–646 (2010).
Macé, A. et al. CNV-association meta-analysis in 191,161 European adults reveals new loci associated with anthropometric traits. Nat. Commun. 8, 744 (2017).
Steele, C. D. et al. Signatures of copy number alterations in human cancer. Nature 606, 984–991 (2022).
Soemedi, R. et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am. J. Hum. Genet. 91, 489–501 (2012).
Marshall, C. R. et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 49, 27–35 (2017).
Bailey, J. A. & Eichler, E. E. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat. Rev. Genet. 7, 552–564 (2006).
Sudmant, P. H. et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015).
Searles Quick, V. B., Davis, J. M., Olincy, A. & Sikela, J. M. DUF1220 copy number is associated with schizophrenia risk and severity: implications for understanding autism and schizophrenia as related diseases. Transl. Psychiatry 5, e697 (2015).
Vollger, M. R., et al. Segmental duplications and their variation in a complete human genome. Science 376, eabj6965 (2022).
Pollen, A. A., Kilik, U., Lowe, C. B. & Camp, J. G. Human-specific genetics: new tools to explore the molecular and cellular basis of human evolution. Nat. Rev. Genet. 24, 687–711 (2023).
Fiddes, I. T. et al. Human-specific NOTCH2NL genes affect Notch signaling and cortical neurogenesis. Cell 173, 1356–1369 (2018).
Vollger, M. R. et al. Increased mutation and gene conversion within human segmental duplications. Nature 617, 325–334 (2023).
Payen, C., Koszul, R., Dujon, B. & Fischer, G. Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet. 4, e1000175 (2008).
Hallast, P., Nagirnaja, L., Margus, T. & Laan, M. Segmental duplications and gene conversion: human luteinizing hormone/chorionic gonadotropin β gene cluster. Genome Res. 15, 1535–1546 (2005).
Hujoel, M. L. A. et al. Protein-altering variants at copy number-variable regions influence diverse human phenotypes. Nat. Genet. 56, 569–578 (2024).
Ho, S. S., Urban, A. E. & Mills, R. E. Structural variation in the sequencing era. Nat. Rev. Genet. 21, 171–189 (2020).
Treangen, T. J. & Salzberg, S. L. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 13, 36–46 (2011).
Liao, W.-W. et al. A draft human pangenome reference. Nature 617, 312–324 (2023).
Ebert, P. et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372, eabf7117 (2021).
Gao, Y. et al. A pangenome reference of 36 Chinese populations. Nature 619, 112–121 (2023).
Wang, T. et al. The Human Pangenome Project: a global resource to map genomic diversity. Nature 604, 437–446 (2022).
Eizenga, J. M. et al. Pangenome graphs. Annu. Rev. Genomics Hum. Genet. 21, 139–162 (2020).
Browning, S. R. & Browning, B. L. Identity by descent between distant relatives: detection and applications. Annu. Rev. Genet. 46, 617–633 (2012).
Chen, C.-H. et al. Determinants of transcription factor regulatory range. Nat. Commun. 11, 2472 (2020).
Wall, J. D. & Pritchard, J. K. Haplotype blocks and linkage disequilibrium in the human genome. Nat. Rev. Genet. 4, 587–597 (2003).
Ogino, S., Gao, S., Leonard, D. G. B., Paessler, M. & Wilson, R. B. Inverse correlation between SMN1 and SMN2 copy numbers: evidence for gene conversion from SMN2 to SMN1. Eur. J. Hum. Genet. 11, 275–277 (2003).
Lorson, C. L., Hahnen, E., Androphy, E. J. & Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl Acad. Sci. USA96, 6307–6311 (1999).
He, Y. et al. T2T-YAO: a telomere-to-telomere assembled diploid reference genome for Han Chinese. Genom. Proteom. Bioinform. 21, 1085–1100 (2023).
Yang, C. et al. The complete and fully-phased diploid genome of a male Han Chinese. Cell Res. 33, 745–761 (2023).
Nurk, S. et al. The complete sequence of a human genome. Science 376, 44–53 (2022).
Handsaker, R. E. et al. Large multiallelic copy number variations in humans. Nat. Genet. 47, 296–303 (2015).
Yilmaz, F. et al. Reconstruction of the human amylase locus reveals ancient duplications seeding modern-day variation. Science 386, eadn0609 (2024).
Bolognini, D. et al. Recurrent evolution and selection shape structural diversity at the amylase locus. Nature 634, 617–625 (2024).
Wong, K. H. Y. et al. Towards a reference genome that captures global genetic diversity. Nat. Commun. 11, 5482 (2020).
Song, L., Bai, G., Liu, X. S., Li, B. & Li, H. Efficient and accurate KIR and HLA genotyping with massively parallel sequencing data. Genome Res. 33, 923–931 (2023).
Numanagić, I. et al. Allelic decomposition and exact genotyping of highly polymorphic and structurally variant genes. Nat. Commun. 9, 828 (2018).
Robinson, J. et al. IPD-IMGT/HLA Database. Nucleic Acids Res. 48, D948–D955 (2020).
Lefranc, M. P. IMGT, the international immunogenetics database. Nucleic Acids Res. 29, 207–209 (2001).
Pacific Biosciences. Pangu https://github.com/PacificBiosciences/pangu (2024).
Wagner, J. et al. Curated variation benchmarks for challenging medically relevant autosomal genes. Nat. Biotechnol. 40, 672–680 (2022).
Prodanov, T. et al. Locityper: targeted genotyping of complex polymorphic genes. Nat. Genet. https://doi.org/10.1038/s41588-025-02362-4 (2025).
Mallick, S. et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538, 201–206 (2016).
Ester, M., Kriegel, H., Sander, J. & Xu, X. A density-based algorithm for discovering clusters in large spatial databases with noise. In KDD’96: Proceedings of the Second International Conference on Knowledge Discovery and Data Mining (eds Simoudis, E. et al.) 226–231 (AAAI, 1996).
Dennis, M. Y., et al. The evolution and population diversity of human-specific segmental duplications. Nat. Ecol. Evol. 1, 69 (2017).
Okada, Y. eLD: entropy-based linkage disequilibrium index between multiallelic sites. Hum. Genome Var. 5, 29 (2018).
Bosch, N. et al. Characterization and evolution of the novel gene family FAM90A in primates originated by multiple duplication and rearrangement events. Hum. Mol. Genet. 16, 2572–2582 (2007).
Lappalainen, T. et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511 (2013).
GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Lan, X. & Pritchard, J. K. Coregulation of tandem duplicate genes slows evolution of subfunctionalization in mammals. Science 352, 1009–1013 (2016).
Keys, K. L., et al. On the cross-population generalizability of gene expression prediction models. PLoS Genet. 16, e1008927 (2020).
Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47, 1091–1098 (2015).
Pajic, P. Independent amylase gene copy number bursts correlate with dietary preferences in mammals. eLife 8, e44628 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Lorson, C. L., Rindt, H. & Shababi, M. Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum. Mol. Genet. 19, R111–R118 (2010).
Hormozdiari, F., Kichaev, G., Yang, W.-Y., Pasaniuc, B. & Eskin, E. Identification of causal genes for complex traits. Bioinformatics 31, i206–i213 (2015).
Tam, V. et al. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484 (2019).
Li, P., Guo, M., Wang, C., Liu, X. & Zou, Q. An overview of SNP interactions in genome-wide association studies. Brief. Funct. Genomics 14, 143–155 (2015).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Ren, J. & Chaisson, M. J. P. lra: a long read aligner for sequences and contigs. PLoS Comput. Biol. 17, e1009078 (2021).
Ross, M. G. et al. Characterizing and measuring bias in sequence data. Genome Biol. 14, R51 (2013).
Kirilenko, B. M., et al. Integrating gene annotation with orthology inference at scale. Science 380, eabn3107 (2023).
Hodson, T. O. Root-mean-square error (RMSE) or mean absolute error (MAE): when to use them or not. Geosci. Model Dev. 15, 5481–5487 (2022).
Daley, T. & Smith, A. D. Predicting the molecular complexity of sequencing libraries. Nat. Methods 10, 325–327 (2013).
Lawson, C. L. & Hanson, R. J. Classics in applied mathematics. in Solving Least Squares Problems 312–337 (SIAM, 1995).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000).
Zhou, Y., Song, L. & Li, H. Full resolution HLA and KIR gene annotations for human genome assemblies. Genome Res. 34, 1931–1941 (2024).
Morales, J. et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature 604, 310–315 (2022).
Mohammadi, P., Castel, S. E., Brown, A. A. & Lappalainen, T. Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Res. 27, 1872–1884 (2017).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Byrska-Bishop, M. et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185, 3426–3440 (2022).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Fox, J. & Weisberg, S. Mixed-effects models in R. in An R Companion to Applied Regression (SAGE, 2002).
Fox, J., Weisberg, S. & Price, B. Car: companion to applied regression. R version 3.1-3 (2019). https://cran.r-project.org/web/packages/car/index.html
Ma, W. & Chaisson, M. Ctyper gene matrices v1.1 [data set]. Zenodo https://doi.org/10.5281/zenodo.16340156 (2025).
Ma, W., & Chaisson, M. Ctyper and analysis source code. Zenodo https://doi.org/10.5281/zenodo.16956833 (2025).
Acknowledgements
This work was supported by NHGRI R01HG011649 (M.J.P.C) and NHGRI U01HG010973 (M.J.P.C.). We thank M. Pennell for helpful critiques of our manuscript.
Author information
Authors and Affiliations
Contributions
W.M. conceived the method, performed the analysis and wrote the paper. M.J.P.C. conceived the method and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Li Song and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–11, Notes 1–12 and mathematical analysis.
Supplementary Tables 1–11 (download XLSX )
Supplemental Tables 1–11.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, W., Chaisson, M.J.P. Genotyping sequence-resolved copy number variation using pangenomes reveals paralog-specific global diversity and expression divergence of duplicated genes. Nat Genet 57, 2909–2919 (2025). https://doi.org/10.1038/s41588-025-02346-4
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41588-025-02346-4
