
Abstract
Intratumor heterogeneity fundamentally challenges cancer treatment, as coexisting, molecularly distinct cell states with non-overlapping drug sensitivities can drive therapeutic resistance. We establish and validate a generalizable, network-based framework to systematically identify combination therapies targeting complementary tumor cell states. Applied to diffuse midline glioma (DMG)—a universally fatal pediatric malignancy—this approach identified master regulator protein dependencies in seven coexisting cell states, confirmed by pooled CRISPR–Cas9 assays. Perturbational transcriptional profiles for 372 clinically relevant drugs prioritized candidates predicted to invert state-specific master regulator activity. State-selective drug sensitivity was validated for eight out of nine (89%) drugs in vivo, including avapritinib, ruxolitinib and larotrectinib. Compared with monotherapy, co-administering drugs targeting complementary states significantly prolonged survival across virtually all combinations, with avapritinib plus ruxolitinib extending median survival nearly threefold versus vehicle and 1.5-fold versus avapritinib alone. These findings establish clinically actionable DMG combinations and a tumor-agnostic and mutation-agnostic framework for rational combination therapy design.
Similar content being viewed by others

Main
The heterogeneity and plasticity of aggressive malignancies represent major barriers to durable treatment response1. Molecularly distinct tumor cell states often exhibit non-overlapping drug sensitivities, including innate resistance. Moreover, cancer cells can spontaneously transdifferentiate, allowing even initially sensitive states to acquire genetically or epigenetically mediated resistance2,3,4. An effective strategy to overcome both innate and adaptive resistance may therefore lie in combinations that, rather than relying on cell-autonomous synergy, target orthogonal dependencies of coexisting cell states.
Single-cell-level tumor profiling represents the key to designing effective drug combinations targeting molecularly distinct cell states. Previous approaches have explored this concept5,6,7, including drug screens in organoids recapitulating cell-state-specific drug sensitivities6, but they typically used small drug panels, relied on in vitro viability and lacked systematic single-cell in vivo validation5,8,9,10. As a result, tumor- and mutation-agnostic identification of effective drug combinations that achieve in vivo synergy, by targeting coexisting cell states, remains a key challenge in precision oncology.
We present a generalizable strategy to achieve this goal by pharmacologically targeting cell-state-specific master regulator (MR) proteins, representing mechanistic determinants of coexisting transcriptional cell states11. MRs represent an actionable class of non-oncogene dependencies that are highly enriched in essential and synthetic lethal proteins11,12,13,14. By leveraging large-scale drug perturbation profiles—capturing the transcriptional responses of cells to clinically relevant drugs—one can identify drugs that abrogate tumor viability in vivo by inverting the activity of either individual essential MRs (OncoTarget15) or of an entire Tumor Checkpoint Module13, a protein module comprising the 50 most differentially activated MRs (OncoTreat16,17). Given that MR activity is assessed from the differential expression of their transcriptional targets12,18,19, this provides a mechanism-based framework to prioritize cell-state-specific drug combinations from single-cell profiles.
Owing to their extensive heterogeneity20,21, DMGs provide an ideal context to test this strategy. These aggressive pediatric brain tumors, characterized by <10% 2-year survival, are driven by oncogenic H3K27-altering histone mutations or EZHIP overexpression—both of which represent non-actionable events—resulting in broad epigenetic dysregulation22,23,24,25,26,27,28,29. Few additional recurrent alterations exist, and resistance has consistently emerged upon their targeting29,30,31,32. These failures reflect the emergence of epigenetically distinct, drug-resistant cell states in the context of the increasingly recognized intrinsic heterogeneity and plasticity of DMGs24,26,29,33,34, and highlight the critical need for rational combination approaches29,35,36.
Here, we systematically identify and pharmacologically target the MR proteins of coexisting DMG cell states. This approach predicted cell-state-specific drug sensitivity with high accuracy, yielding multiple drug combinations that significantly outperformed their associated monotherapies in vivo. This procedure culminated in the identification of clinically actionable combinations poised for rapid clinical translation, including avapritinib/ruxolitinib and avapritinib/larotrectinib. More broadly, these findings establish a generalizable framework for designing combination therapies targeting coexisting cell states in heterogeneous tumors.
Results
Network-based analysis elucidates MRs controlling DMG cell states
Using metaVIPER37, the single-cell extension of the VIPER algorithm38, we inferred the activity of transcriptional regulators based on differential expression of their DMG-specific regulatory targets, as inferred by ARACNe18,19,39. Analyses were performed on 3,032 malignant single cells profiled by single-cell RNA sequencing (scRNA-seq) from six DMG tumors20, following removal of non-malignant microglial cells by inferCNV analysis40 (Supplementary Fig. 1a–c,e). This approach is robust to gene dropout, enabling activity inference even for proteins whose encoding genes cannot be detected41.
Resolution-optimized Louvain clustering of VIPER-inferred protein activity (Supplementary Fig. 1f) revealed seven malignant cell states, conserved across each tumor, thus further refining the prior four-state classification20 (Fig. 1a,b and Supplementary Fig. 1g). State identity was defined by established glial lineage marker enrichment among the top 50 most differentially activated proteins of each state (cell state MRs; Fig. 1c,d). Based on OLIG1/OLIG2/PDGFRA activity, three oligodendrocyte precursor cell (OPC)-like states were identified and termed quiescent (OPCQ), canonical (OPC) and cycling (OPCC) based on their proliferative signatures. Additionally, based on SIRT2/PLP1/MPZL1 and SOX9/APOE/HEY1 activity, two oligodendrocyte (OC)-like states (OC and OPC/OC) and two astrocyte (AC)-like states (AC and OPC/AC) were identified, respectively, with each pair representing stable and transitional cell states. OPC states comprised the majority of malignant cells (Supplementary Fig. 1g).

a, UMAP projection of VIPER-inferred protein activity across 3,032 tumor cells from six patients with DMG, with cells colored by seven clusters identified using resolution-optimized Louvain clustering. b, Heatmap of single-cell protein activity (NES) showing the top 15 most activated MRs per cluster (cell state MRs). Protein activity is represented as a color gradient from blue (aberrantly inactivated) to red (aberrantly activated). Cluster assignment and patient identity for each single cell are indicated in the annotation bars above the heatmap. Cells from all six patients contribute to each of the seven clusters. c, Heatmap of Stouffer-integrated protein activity (NES) for established OPC, OC and AC lineage markers, as well as proliferative markers, across clusters. Marker activity patterns supported cluster annotation by glial lineage and proliferation status (cluster identities shown along the x axis). d, UMAP from a colored by inferred tumor cell state annotation, used for downstream comparisons between patient-derived cells and tumor model systems. e, Heatmap showing pathway enrichment across tumor cell states, expressed as −log10(FDR-adjusted P values), based on overrepresentation analysis of cluster-specific differentially expressed genes in MSigDB Hallmark 2020 gene sets using a one-sided Fisher’s exact test. f, Schematic of pooled CRISPR–Cas9 knockout screens performed in three patient-derived DMG cell lines to assess the essentiality of VIPER-inferred tumorigenic MRs. TFs, transcription factors. Illustrations created in BioRender; Calvo Fernandez, E. https://BioRender.com/kyxr92e (2025). g, One-sided GSEA showing significant enrichment (P = 6.8 × 10−3) of DMG CRISPR-essential genes (red ticks) within the VIPER-inferred synthetic bulk tumorigenic protein activity signature of all DMG malignant cells (GTEx caudate reference). h, Heatmap of cell-state-specific synthetic bulk tumorigenic protein activity for 45 CRISPR-validated essential MRs (rows) comprising the leading edge of enrichment in at least one DMG cell state (columns). The annotation bar indicates state-specific enrichment of DMG-essential genes based on one-sided GSEA, with black denoting significant enrichment (FDR ≤ 0.05) and gray denoting non-significance.
Pathway enrichment of state-specific differentially expressed genes (Supplementary Table 1) revealed targetable and developmentally related programs (Fig. 1e). OPC cells showed dysregulation of proliferation and OPC-to-OC differentiation pathways (e.g. ‘MYC targets’42, ‘Notch signaling’43,44,45, and ‘Wnt/β catenin signaling’44). OC-like cells uniquely activated ‘mTORC1 signaling’46,47, and OPC/AC-like cells showed activation of ‘IL-6–JAK–STAT3 signaling’, promoting angiogenesis48, reactive astrogliosis49,50 and impaired OPC-to-OC differentiation51.
These findings—including cell states and associated MRs—were reproduced in an independent scRNA-seq dataset from eight pediatric DMGs21 (Extended Data Fig. 1a–d). Integrative analyses confirmed the highly recurrent nature of the seven cell states, with all samples showing significant cell-state-specific MR conservation, independent of their mutational repertoire (Extended Data Fig. 1e–g).
CRISPR screens validate tumorigenic MR dependencies
To further assess whether MRs driving the ‘normal-to-tumor’ transition represent functional DMG dependencies, we inferred protein activity by VIPER analysis of genes differentially expressed in malignant synthetic bulk profiles—that is, generated by adding the RNA-seq reads of all single cells—versus GTEx normal caudate samples (tumorigenic signature). We then assessed enrichment of candidate tumorigenic MRs; that is, the 50 most aberrantly active proteins, in DMG-essential genes, as identified by pooled CRISPR–Cas9-mediated knockout screens targeting all transcription factors. These were performed in three genetically distinct DMG cell lines: SF8628 (H3F3A K27M), DIPG4 (HIST1H3B K27M, TP53Mut, ACVR1Mut) and DIPG17 (H3F3A K27M, TP53Mut) (Fig. 1f, Extended Data Fig. 2f and Supplementary Table 3).
Aberrantly active MRs were significantly enriched in DMG-essential genes as defined by log2(fold change) ≤0 and false discovery rate (FDR) ≤0.05 (P = 6.8 × 10−3, one-sided gene set enrichment analysis (GSEA) integrated across all cell lines; Fig. 1g and Extended Data Fig. 2a). As expected, enrichment was strongest for MRs of cell states comprising the majority of cells in each cell line (that is, OPC and OPCC; Fig. 1h and Extended Data Fig. 2b,g). Among 45 essential MRs overall—in the leading edge of at least one cell-state-specific enrichment analysis (Supplementary Fig. 2b)—Forkhead box M1 (FOXM1) emerged as the most conserved dependency (Fig. 1h and Extended Data Fig. 2d)52. Several additional essential MRs (for example, DLX1, ZZZ3, GMEB1, DLX2, TGIF2, SOX10) represent established H3K27me3 targets53,54, suggesting epigenetic de-repression by H3K27M as a potential activation mechanism.
Although most essential MRs were conserved across tumors, VIPER analysis also captured a small repertoire of sample-specific and mutation-specific dependencies. For example, essential genes in DIPG4—uniquely harboring the HIST1H3B mutation—were only significantly enriched in MR dependencies from the MUV5 sample harboring the same mutation (Extended Data Fig. 2c). Pontine-derived samples showed consistently stronger enrichment than thalamic tumors, aligned with the anatomic origin of the cell lines. Moreover, when enrichment was assessed between cell-line-specific MRs (assessed by bulk RNA-seq) and matched cell-line-specific dependencies, statistical significance increased by two orders of magnitude (Extended Data Fig. 2e). These results highlight the ability of VIPER to nominate sample-specific and mutation-specific MRs and underscore the presence of idiosyncratic cell-line-specific dependencies.
Together, these data validate that VIPER-inferred tumorigenic MRs are enriched in functional DMG dependencies, especially for the predominant OPC states, thus supporting in vivo testing of MR-targeting drugs (MR-inverters), including for the minority states (that is, OC and AC).
Drug perturbations define DMG-specific mechanism of action
To identify MR-inverter drugs for each cell state, we profiled the transcriptional responses of two DMG cell lines, SF8628 (H3F3A K27M) and DIPG6 (H3F3A K27M, TP53Mut), following treatment with 372 US Food and Drug Administration (FDA)-approved and late-stage oncology compounds (Fig. 2a and Supplementary Table 4). These were selected based on MR conservation analysis (OncoMatch55), which confirmed their strong complementary concordance with tumorigenic MRs across bulk DMG samples and the seven malignant cell states. Specifically, DIPG6 and SF8628 emerged as effective models for OPC/OC and AC states, respectively (FDR ≤ 10−30 and FDR ≤ 10−20, one-sided analytic rank-based enrichment analysis (aREA); Fig. 2b).

a, Schematic of the experimental and computational workflow used to characterize drug-induced differential protein activity in DMG. Using PLATE-seq, RNA-seq profiles were generated 24 h after treatment of two DMG cell lines selected by OncoMatch to recapitulate human DMG tumorigenic MRs. Cells were treated with 372 FDA-approved and late-stage investigational oncology compounds. VIPER was used to infer drug-induced differential protein activity relative to DMSO controls, enabling nomination of drugs capable of inverting DMG cell-state-specific tumorigenic MRs. Illustrations created in BioRender; Calvo Fernandez, E. https://BioRender.com/cbt4zn0 (2025). b, OncoMatch analysis showing enrichment of synthetic bulk tumorigenic MRs from each DMG cell state (columns; GTEx caudate reference) within the VIPER-inferred tumorigenic protein activity signature of six human DMG cell lines (rows; GTEx caudate reference). Values represent −log10(FDR-adjusted P values) derived from one-sided aREA. c,d, Heatmaps depicting 24 h drug-induced protein activity profiles for SF8628 (c) and DIPG6 (d) cells across a representative subset of drugs profiled by PLATE-seq. Rows correspond to drugs and columns to the top 50 most aberrantly active tumorigenic MRs for each of the seven DMG cell states. Color scale denotes VIPER-inferred differential protein activity (NES) comparing drug-treated versus DMSO-treated cells, from blue (inactivated) to red (activated). Drugs are annotated by previously reported MoA and grouped by unsupervised hierarchical clustering of their protein activity signatures. Tumorigenic MRs are grouped by the DMG cell state(s) in which they were most differentially active and by directionality (most activated (↑) or most inactivated (↓) within each state).
VIPER analysis of drug versus vehicle control-treated cells using PLATE-seq56 bulk RNA-seq profiles collected at 24 h post treatment produced proteome-wide activity signatures representing the DMG-specific MoA of each drug16,17,57 (Fig. 2a).
Hierarchical clustering of MoA profiles (Fig. 2c,d) showed that drugs with related high-affinity targets often clustered together, whereas others with distinct targets converged on highly similar MoA profiles, reflecting DMG context-specific canalization. Drug clusters also exhibited distinct patterns of tumorigenic MR activity modulation across DMG cell states, providing a mechanistic basis for identifying state-selective MR-inverter compounds based on negative enrichment (that is, activity reversal) of tumorigenic MRs among proteins differentially activated by each drug (OncoTreat algorithm).
Predicted MR-inverter drugs segregate with DMG cell states
To identify drug combinations targeting each coexisting, molecularly distinct DMG cell state, we first identified candidate MRs by VIPER analysis of genes differentially expressed in each cell state versus GTEx normal caudate samples. Then, we identified established high-affinity inhibitors of individual MRs (OncoTarget) and drugs inverting the activity of the top 50 tumorigenic MRs, based on SF8628 and DIPG6 perturbational profile analysis (OncoTreat; Fig. 2a and Supplementary Table 4). Unlike conventional in vitro drug screens, this mechanism-based approach focuses on vulnerabilities controlling each cell state, as assessed from primary tumors, rather than by empirical cell-line-based viability assays.
Predicted MR-inverter drugs co-segregated strongly with OPC, AC and OC states, highlighting their distinct therapeutic vulnerabilities (Fig. 3a). To select candidates for in vivo validation, we prioritized drugs with (1) Bonferroni-adjusted one-sided MR P value (P ≤ 10−5, see prior work17), (2) physiologically relevant perturbation concentrations, (3) multi-state or highly state-specific predicted sensitivity and (4) concordant OncoTarget and OncoTreat prediction. Pediatric clinical relevance, bioavailability and blood–brain barrier (BBB) permeability were also considered.

a, Heatmaps showing the statistical significance of OncoTarget-predicted drug targets (left; rows) and OncoTreat-predicted MR-inverter drugs (right; rows) across the seven DMG cell states (columns) quantified as −log10(Bonferroni-adjusted P values) derived from the respective one-sided aREA, displayed both as a maroon color scale and numerically within each heatmap cell. Analyses were performed using cell-state-specific, Stouffer-integrated synthetic bulk VIPER-inferred tumorigenic protein activity profiles (GTEx caudate reference). The OncoTarget Top and OncoTreat Top heatmaps display the top 20 drug targets and MR-inverter drugs (that is, drugs predicted to invert the activity of the top 50 most aberrantly active tumorigenic MRs), respectively, for each cell state. The OncoTarget Unique heatmap highlights significantly activated drug targets uniquely associated with individual cell states. b, Schematic illustrating the known MoA of five drugs selected to target OPC-like states (purple) and four drugs selected to target AC-like states (pink) for in vivo validation. Figure adapted from previous publications83,84 and created in BioRender; Calvo Fernandez, E. https://BioRender.com/ud8gakw (2025).
Using these criteria, OPC state analysis predicted trametinib (MEK1/2)58, mocetinostat (HDAC)59 and dinaciclib (CDK1/2/5/9)60 as OncoTreat MR-inverters, while OncoTarget identified etoposide (TOP2)61 and avapritinib (PDGFRA)62. For AC states, OncoTreat predicted venetoclax (BCL2)63 and ruxolitinib (JAK1/2)63, while OncoTarget predicted larotrectinib (NTRK)64 and napabucasin (STAT3)65. These agents either ranked among the top state-specific predictions or were predicted across multiple states, thus supporting the evaluation of both broad and cell-state-specific drugs. Selected drugs and their high-affinity targets are summarized in Fig. 3b. Additional OC-specific drugs (for example, temsirolimus, ipatasertib) may be evaluated in future studies.
DMG heterogeneity is recapitulated in vivo but not in vitro
To identify which models preserve human DMG cell state diversity, we analyzed scRNA-seq from in vitro and in vivo DMG models. Cell state assignment was based on state-specific MR enrichment by GSEA in single-cell protein activity signatures (Supplementary Table 1). For simplicity, cells enriched for OPCQ/OPC MRs were labeled ‘OPC’, those enriched for OPC/OC or OC MRs as ‘OC’ and those enriched for OPCC, AC or OPC/AC MRs as the corresponding state.
Although tumorigenic MRs, especially for OPC states, were conserved in cell line bulk profiles (Fig. 2b), single-cell analysis of six DMG cell lines (n = 11,325 cells; Extended Data Fig. 2f) revealed that 79.4% of cells did not significantly match any state, suggesting substantial in vitro adaptation and drift. Remaining cells were classified as OPCC, consistent with the proliferative bias of cell culture (Extended Data Fig. 2g). Therefore, in vitro culture abrogates the native DMG cell state architecture, with protein activity likely reflecting ex vivo culture conditions.
We next evaluated whether in vivo passaging could restore human tumor heterogeneity. Across 14,176 tumor cells from a DIPG17 (H3F3A K27M)-derived subcutaneous xenograft model (Fig. 4a and Supplementary Fig. 3a,b), VIPER-based clustering recapitulated human cell state heterogeneity, with strong concordance between unsupervised clustering and cell-state assignments, as inferred by statistically significant enrichment of state-specific MRs (Fig. 4b and Supplementary Fig. 3e). Like human tumors, OPC cells predominated (66.2%), but AC (27.0%) and OC (2.2%) cells were also detected (Fig. 4b and Supplementary Fig. 3f). Only 4.6% of cells remained unclassified, and both cell-state MRs and state-specific drug predictions were significantly conserved between mouse and human tumors (Fig. 4c,d). This confirms that in vivo models, unlike in vitro cultures, faithfully recapitulate DMG cell state architecture for preclinical testing of state-specific drug efficacy.

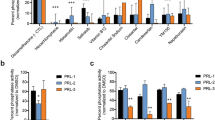
a, Bioluminescence imaging 1 month post-injection, confirming tumor implantation in the DIPG17 subcutaneous xenograft model. b, UMAP projection of VIPER-inferred protein activity from 14,176 single cells across four vehicle-treated tumors, colored by assigned human DMG cell state based on the strongest statistically significant enrichment of human state-specific MRs per cell (one-sided GSEA, FDR-adjusted). c, Heatmap of viperSimilarity NES quantifying concordance between human and mouse cell state MRs. Red indicates concordant enrichment and blue indicates inverse enrichment. d, OncoTarget and OncoTreat scores for drugs tested in vivo, shown as −log10(Bonferroni-adjusted P values) derived from one-sided aREA, demonstrating conservation of predicted cell-state-specific activity between human tumors and the model. e, Percent change in tumor volume 5 days post treatment across nine drug arms and vehicle control. Statistical significance between drug-treated and vehicle-treated tumors was assessed using two-sided Welch’s t-test. Representative ultrasound images before and after avapritinib treatment illustrate treatment-associated morphological changes (pink arrow). Each data point represents an individual mouse (biological replicate): vehicle (n = 5), trametinib (n = 4), mocetinostat (n = 3), napabucasin (n = 3), avapritinib (n = 2), dinaciclib (n = 2), etoposide (n = 2), larotrectinib (n = 2), ruxolitinib (n = 2) and venetoclax (n = 2). f,g, log2(fold change (FC)) in the fraction of cells assigned to each cell state following drug treatment relative to vehicle-treated tumors (14,176 single cells from four tumors), across 38,615 single cells from tumors treated with five OPC-targeting drugs (f) and 42,896 single cells from tumors treated with four AC-targeting drugs (g). Bar heights represent mean log2FC across biological replicates (each dot represents an independent tumor); error bars, s.e.m. Statistical significance of differences in the proportion of cells assigned to the predicted targeted cell state was assessed using χ2 tests for each treatment arm, with FDR correction applied within each drug across cell states: avapritinib (n = 2; POPC < 10−300), mocetinostat (n = 2; POPC = 6.94 × 10−298), dinaciclib (n = 2; POPC < 10−300), etoposide (n = 2; POPC < 10−300), trametinib (n = 2; POPC < 10−300); ruxolitinib (n = 2; PAC = 3.36 × 10−6), larotrectinib (n = 2; PAC = 6.14 × 10−13), venetoclax (n = 3; PAC = 4.64 × 10−2) and napabucasin (n = 3; PAC = 2.51 × 10−15; significant shift opposite to the predicted direction). Asterisks denote statistical significance: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. h, Pharmacodynamic analysis of cell-state-specific tumorigenic MR activity inversion in vivo. Analytic aREA plots show enrichment of the top (red ticks) and bottom (blue ticks) 50 human DMG cell-state-specific tumorigenic MRs in drug-treated versus vehicle-treated protein activity signatures from the same cell state. Representative examples are shown: trametinib in OPC-like cells and venetoclax in AC-like cells. P values correspond to two-sided aREA for each individual drug-state comparison. i, Cell-state-specific differential protein activity analyses (NES, x axis) confirming cell-state-specific (y axis) drug-mediated inversion of OncoTarget-predicted dependencies: CDK2 activity inhibition by dinaciclib in OPC-like cells (P = 5.1 × 10−33, one-sided aREA) and JAK1 inhibition by ruxolitinib in AC-like cells (P = 1.8 × 10−9, one-sided aREA).
Drugs deplete predicted cell states in DIPG17 xenografts
We next tested whether predicted MR-inverter drugs selectively depleted their target cell states in vivo, using the DIPG17 subcutaneous xenografts. This process was designed to assess state-specific pharmacodynamic effects and cell state depletion independent of BBB permeability, as a pre-screen for validation in orthotopic models. Tumor-bearing mice were treated for 5 days, tumor volume was monitored by ultrasound and tumors were dissected after the final dose for scRNA-seq (Supplementary Fig. 3a,b). For 95,687 single cells from 24 samples across nine drug arms (n = 2 or 3 per arm) and vehicle control (n = 4), cell states were assigned based on state-specific MR enrichment by GSEA in single-cell protein activity signatures, and differential state occupancy in drug-treated versus vehicle-treated animals was assessed by χ2 analysis (Supplementary Fig. 3d,f and Supplementary Table 5).
We validated selective cell state depletion consistent with predictions for eight out of nine tested drugs (Fig. 4f,g). All five OPC-targeting drugs significantly and specifically depleted OPC (OPCQ/OPC) and OPCC states, including trametinib (POPC = 2.22 × 10−91, POPCC = 4.46 × 10−144), dinaciclib (POPC = 2.52 × 10−136, POPCC = 5.81 × 10−121), mocetinostat (POPC = 4.24 × 10−320, POPCC = 5.43 × 10−03), etoposide (POPC = 3.77 × 10−247, POPCC = 5.95 × 10−20) and avapritinib (POPC = 2.71 × 10−201, POPCC = 4.84 × 10−56) (Fig. 4f and Supplementary Fig. 4a). As anticipated, other states increased in relative abundance, particularly AC states (PAC < 10−300, PAC = 9.39 × 10−147, PAC = 5.34 × 10−161, PAC = 7.71 × 10−89 and PAC = 2.79 × 10−82, respectively) and, in some cases, OC states, especially with avapritinib (POC < 10−300) and etoposide (POC = 4.29 × 10−219), indicating innate resistance to monotherapies.
Three of the four AC-targeting drugs significantly depleted AC and/or OPC/AC states, including ruxolitinib (PAC = 2.38 × 10−8, POPC/AC = 0.18), venetoclax (PAC = 6.53 × 10−08, POPC/AC = 8.50 × 10−11) and larotrectinib (PAC = 8.21 × 10−03, POPC/AC = 3.41 × 10−35) (Fig. 4g and Supplementary Fig. 4b). Effects on OPC and OC states were consistent with model-specific predictions. Ruxolitinib showed a significant relative increase in OPCC cells (POPCC = 1.04 × 10−9). Venetoclax, predicted to also target OPCC cells in this model, effectively depleted them (POPCC = 1.33 × 10−57), with a significant relative increase in OC cells (P < 10−300). Larotrectinib uniquely and significantly depleted OC cells (POC = 1.50 × 10−26). Although predicted to target AC states, napabucasin significantly depleted only the OPC state (POPC = 2.89 × 10−192).
As expected from drugs targeting individual states, tumor volume measurements showed only modest growth reductions and only for drugs targeting the dominant OPC states, most pronounced for dinaciclib and avapritinib, with additional activity observed for ruxolitinib and napabucasin (Fig. 4e).
Tumorigenic MR activity inversion is conserved in vivo
To confirm that predicted drugs preserve their in-vitro-derived MoA in vivo, we evaluated whether they inverted state-specific tumorigenic MR activity in PDX models. All OncoTreat-predicted drugs—mocetinostat, trametinib and dinaciclib (OPC-targeting), and venetoclax and ruxolitinib (AC-targeting)—significantly inverted their respective state-specific tumorigenic MRs (P≤ 0.05, one-sided aREA; Fig. 4h and Extended Data Figs. 3 and 4a). Ruxolitinib showed borderline inversion of AC MRs (P = 0.058) but significantly inhibited JAK1 (P = 1.8 × 10−9), supporting its OncoTarget-based prioritization (Fig. 4i). Avapritinib induced broad inversion of OPC state MRs (Extended Data Fig. 3). Larotrectinib inhibited NTRK2 in OC and especially OPC/AC cells (P = 0.03) (Extended Data Fig. 4b) despite limited baseline NTRK2 activity, suggesting that the inferred regulon may inadequately capture functional target activity. The only exception was napabucasin, which failed to inhibit STAT3 in vivo, providing a potential rationale for its inability to elicit its predicted AC cell sensitivity (Extended Data Fig. 4c).
Monotherapies targeting dominant states improve survival
To validate drug efficacy within a native tumor location and microenvironment, we tested predicted drugs in an immunocompetent orthotopic DMG model66 using DIPG4423 (Pdgfb+, Trp53−/−, H3f3a K27M) murine cells. Magnetic resonance imaging (MRI) and histology confirmed pontine engraftment, and scRNA-seq of untreated tumors (n = 8,013 cells) recapitulated human DMG cell states and drug predictions, supporting model relevance (Fig. 5a–e and Extended Data Fig. 5a–g). Following engraftment, mice were treated with each of the nine drugs (n = 3–5 per arm) or vehicle (n = 9), and responses were assessed by MRI tumor volume at 2 weeks and for overall survival (Extended Data Fig. 6a,b and Supplementary Fig. 5a,b).

a, Schematic of stereotactic injection coordinates for establishment of the DIPG4423 syngeneic orthotopic DMG murine model (top). Tumor cells were injected 1 mm inferior and lateral to the lambda suture (purple circle; gray dot indicates injection site). Representative sagittal hematoxylin and eosin (H&E) staining confirms accurate tumor localization within the pons (bottom). b,c, Representative H&E images of normal brain parenchyma (b) and tumor periphery (c), showing diffuse infiltration of tumor cells into adjacent normal tissue. H&E staining was performed for all mice included in survival analyses, when feasible. d, UMAP projection of VIPER-inferred protein activity from 8,013 tumor single cells across two untreated DIPG4423 pontine tumors. Cells are colored by assigned human DMG cell state based on the strongest enrichment of human state-specific MRs per cell (one-sided GSEA, FDR-adjusted). e, OncoTarget and OncoTreat scores for drugs tested in this model, shown as −log10(Bonferroni-adjusted P values) derived from one-sided aREA, demonstrating conservation of predicted cell-state-specific activity between human tumors and the model. f, Representative MRI of tumors 37 days post treatment with monotherapies that significantly extended survival, compared with vehicle-treated controls imaged at 25 days. g, High-power H&E images of end-stage tumors from vehicle-treated, trametinib-treated and avapritinib-treated mice, showing diffuse infiltration with minimal necrosis in avapritinib-treated tumors. h, Tumor volume measured 2 weeks post treatment across independent biological replicates treated with OPC-targeting drugs (left) or AC-targeting drugs (right). OPC-targeting drugs included avapritinib (n = 4), dinaciclib (n = 5), mocetinostat (n = 5), trametinib (n = 5) and etoposide (n = 3). AC-targeting drugs included ruxolitinib (n = 4), larotrectinib (n = 3), venetoclax (n = 5) and napabucasin (n = 3). Drugs are ranked by statistical significance relative to vehicle control (n = 9; two-sided Welch’s t-test). i, Kaplan–Meier survival analysis of monotherapy arms demonstrating significant survival benefit compared with vehicle control (log-rank test): avapritinib (P = 1.9 × 10−3), dinaciclib (P = 3.3 × 10−3) and trametinib (P = 0.03). j, Representative MRI images 37 days post treatment show reduced tumor burden following treatment with OPC-targeting and AC-targeting drug combinations compared with vehicle or corresponding monotherapies from f. k, Tumor volume 2 weeks post treatment for avapritinib (n = 5), trametinib (n = 5) and dinaciclib (n = 5), administered alone or in combination with AC-targeting drugs, including avapritinib + venetoclax (n = 5), avapritinib + ruxolitinib (n = 3), avapritinib + larotrectinib (n = 3), trametinib + ruxolitinib (n = 4), trametinib + venetoclax (n = 5) and dinaciclib + ruxolitinib (n = 3). Statistical significance between combination therapies and their respective monotherapies was assessed using two-sided Welch’s t-test. l, Kaplan–Meier survival analysis comparing vehicle, monotherapy and combination therapy arms for avapritinib, trametinib and dinaciclib. Four out of six combinations significantly improved survival relative to the corresponding monotherapy (log-rank test): avapritinib + ruxolitinib (P = 0.02), trametinib + ruxolitinib (P = 0.01), dinaciclib + ruxolitinib (P = 0.01) and avapritinib + larotrectinib (P = 0.02). Asterisks denote statistical significance: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; n.s., not significant. Panel a created in BioRender; Calvo Fernandez, E. https://biorender.com/nhid41b (2026).
OPC-targeting drugs significantly impaired tumor growth (P ≤ 0.05, two-sided Welch’s t-test), except etoposide, probably owing to limited BBB penetration (Fig. 5f,h and Extended Data Fig. 6a). Avapritinib, dinaciclib and trametinib also extended survival (median of 53.5 days, 30 days and 28 days, respectively, versus 25 days for vehicle; P ≤ 0.05, log-rank test; Fig. 5i). Among AC-targeting drugs, only ruxolitinib modestly impaired tumor growth (P = 1.4 × 10−3, two-sided Welch’s t-test), and, as expected for therapies targeting a minority state, none improved survival (Fig. 5h and Supplementary Fig. 5b). MRI and postmortem histology revealed that avapritinib-treated tumors were more diffusely infiltrative and lacked necrosis, consistent with phenotypes previously reported with PDGFR inhibition67 (Fig. 5f,g, Extended Data Fig. 6a and Supplementary Fig. 6).
Combinations targeting complementary states enhance efficacy
To evaluate whether co-targeting complementary cell states enhances tumor control, we treated mice (n = 3–5 per arm) with drug pairs shown to target OPC and AC states, which are the most abundant complementary populations. OPC-targeting drugs (avapritinib, dinaciclib, trametinib) were selected based on monotherapy survival benefit, and AC-targeting drugs (ruxolitinib, venetoclax) were selected based on tumor growth inhibition. We also tested avapritinib/larotrectinib, the only combination with an OC-depleting drug. All combinations were tolerated except dinaciclib/venetoclax, which caused early weight loss.
All combinations significantly reduced tumor growth compared to vehicle (Extended Data Fig. 7b), and several outperformed their associated monotherapies based on a two-sided Welch’s t-test, including avapritinib/venetoclax (P = 0.01), trametinib/ruxolitinib (P = 0.02) and dinaciclib/ruxolitinib (P = 0.046); avapritinib/ruxolitinib trended similarly (P = 0.068) (Fig. 5j,k and Extended Data Figs. 7–9).
More relevant, a greater number of combinations significantly improved median survival (log-rank test) compared to monotherapy, including all ruxolitinib combinations: avapritinib/ruxolitinib (83 days vs 53.5 days; P = 0.02), trametinib/ruxolitinib (45.5 days vs 28 days; P = 0.01) and dinaciclib/ruxolitinib (48 days vs 30 days; P = 0.01), as well as avapritinib/larotrectinib (67 days vs 53.5 days; P = 0.02) (Fig. 5l). Given that ruxolitinib and larotrectinib were ineffective alone, the combinations are by definition synergistic.
Avapritinib/ruxolitinib extended survival despite only modest early volume reduction, suggesting sustained suppression of adaptive resistance owing to selection of a minority cell state. By contrast, avapritinib/venetoclax impaired tumor growth but did not extend survival, possibly related to a more infiltrative phenotype with seizure-related deaths (Supplementary Fig. 7).
Tumor synergy occurs without cell-autonomous synergy
To determine whether avapritinib/ruxolitinib synergy reflects targeting of distinct cell states rather than cell-autonomous synthetic lethality mechanisms, we performed BLISS independence analyses68 in three OPC-predominant DMG cell lines (DIPG4423, SF8628, DIPG36). Given that Bliss synergy requires a combined effect exceeding the sum of individual effects, synergy driven by independent activity in distinct states should disappear in cultures dominated by a single state. Indeed, all lines showed additive, not synergistic, interactions (Extended Data Fig. 10a), indicating that the in vivo synergy would be missed by traditional in vitro assays. Consistent with this interpretation, monotherapy dose–response curves showed robust avapritinib activity but minimal effect of ruxolitinib, with DIPG36 cells even expanding during treatment (Extended Data Fig. 10b,c). These findings support that the efficacy of ruxolitinib is specific to the AC state, which is largely absent in vitro.
Validation of bulk RNA-seq predictions for precision oncology
Given that single-cell protein activity analyses identified effective combination therapies for DMG, we asked whether predictions from more accessible bulk RNA-seq profiles could similarly guide treatment selection for patients. Although potentially biased toward dominant OPC states, they may capture inter-patient heterogeneity and enable broader clinical application.
We analyzed samples from three patients with DMG (MTB36, MTB37, MTB41) whose tumors underwent bulk RNA-seq and in vitro drug screening through a precision medicine program at Rady Children’s Hospital69. MTB36 and MTB37 were screened with 63 drugs (52 in PLATE-seq), and MTB41 was screened with 175 drugs (124 in PLATE-seq), using IC50 and CellTiter-Glo viability readouts. For each sample, OncoTarget/OncoTreat was used to predict candidate drugs expected to invert sample-specific MR activity (Fig. 6a).

a, Heatmaps showing the statistical significance of OncoTarget-predicted drug targets, OncoTreat-predicted MR-inverter drugs and OncoMatch-inferred cell line model similarity for three patient-derived DMG bulk tumor RNA-seq samples (MTB36, MTB37, MTB41). Significance is reported as −log10(Bonferroni-adjusted P values) derived from the respective one-sided aREA, displayed both as a maroon color scale and numerically within each heatmap cell. Analyses were performed using VIPER-inferred tumorigenic protein activity signatures computed relative to the GTEx caudate reference. b, Scatter plot showing the relationship between OncoTreat score and in vitro drug sensitivity across 223 drugs profiled by both bulk RNA-seq and functional drug screens. The x axis represents the negative of the OncoTreat NES (−NES), such that higher values indicate stronger predicted inversion of tumorigenic MRs, and the y axis represents in vitro log(IC50). A two-sided Spearman rank correlation is shown (ρ = −0.14, P = 0.039). Gray and red vertical dashed lines denote OncoTreat score thresholds corresponding to Bonferroni-adjusted P values of 0.01 and 1 × 10−5, respectively. The horizontal red dashed line indicates IC50 ≈ 10 μM. Points in the lower right quadrant correspond to true positive predictions, defined as OncoTreat-predicted drugs with confirmed in vitro activity. c, Heatmap summarizing in vitro validation of OncoTarget/OncoTreat-predicted drugs (Bonferroni-adjusted P value of ≤0.01, one-sided aREA). Drug efficacy was defined by IC50 ≤ 10 μM and viability of ≤50%. Predictive performance was evaluated based on specificity and positive predictive value. Cell colors indicate prediction outcomes: true positives (TP), true negatives (TN), false negatives (FN) and false positives (FP). Numerical values within each cell denote the statistical significance as −log10(Bonferroni-adjusted P value) of the OncoTreat prediction for each drug (row) in each patient sample (column).
Among 228 drugs tested by both approaches, OncoTarget/OncoTreat predictions (P ≤ 0.01, Bonferroni-corrected one-sided aREA) were twice as likely to be effective (IC50 ≤ 10 μM and viability of ≤50%) in the matched patient as non-predicted drugs (odds ratio, 2.28; 95% CI, 1.21–4.41; two-sided P = 0.0076). Predictions showed high specificity, correctly excluding 80.5% (91 out of 113) of inactive drugs, and a strong positive predictive value, with 65.1% (41 out of 63) of predicted drugs proving effective (Fig. 6c). Overall accuracy was 57.9% (132 out of 228), with significant correlation between OncoTreat score and IC50 (Spearman’s p = 0.039) (Fig. 6b).
These findings support the use of bulk RNA-seq profile analyses to prioritize patient-specific therapies and provide a mechanistic rationale for clinical trial design. Critically, as MRs are inferred from patient tumors, not cell culture, in vitro assays may underrepresent their full effect (for example, on migration or immunosuppression), consistent with prior work showing even greater efficacy in vivo17.
Discussion
The rational design of combination therapies remains a major challenge, as most approaches rely on synthetic lethality screens or empirical testing, limiting in vivo translation and generalizability. An alternative strategy is to leverage single-cell analyses to target coexisting tumor cell states with non-overlapping drug sensitivities. Although conceptually promising5,6,8,9,10, previous efforts were limited by small drug panels and a lack of systematic in vivo validation at single-cell resolution.
This study presents and validates a generalizable, mechanism-based framework for rational combination therapy design that (1) identifies MR proteins as essential drivers of molecularly distinct tumor cell states, (2) predicts clinically relevant MR-inverter drugs inducing targeted cell state depletion with nearly 90% accuracy and (3) nominates combinations targeting complementary cell state dependencies that synergize in vivo.
An important distinction of this strategy is that it does not rely on in vitro drug sensitivity, which lacks context-specific mechanistic insight and is biased by cell-line-specific dependencies. Instead, we use in vitro drug-induced transcriptional profiles solely to define proteome-wide MoA, enabling prioritization of drugs that invert human state-specific tumorigenic MRs. Prior studies using bulk RNA-seq have shown highly predictive MR activity inversion translating to in vivo responses12,14,15,16. Extending this strategy to single-cell resolution enables rational combination design for heterogeneous tumors and provides a mechanism-based approach to address both innate and adaptive resistance.
Although broadly applicable, we focused this strategy on DMG, a universally fatal pediatric brain tumor characterized by global epigenetic dysregulation, heterogeneous co-occurring somatic mutations (that is, TP53, ACVR1, EGFR)21,22,23,24,25 and marked intratumor heterogeneity20,21 that has hindered development of targeted therapies. Using metaVIPER, we identified seven recurrent DMG cell states across 14 tumors, each driven by conserved MR programs. Protein activity-based analysis resolved discrete and transitional states more clearly than gene expression alone, consistent with evidence that oncogenic programs converge onto shared, mutation-agnostic regulatory modules13. CRISPR–Cas9 screens in genetically distinct DMG cell lines confirmed significant enrichment of VIPER-inferred MRs among tumor-essential genes, demonstrating their functional relevance.
This study extends prior work describing intratumor heterogeneity in DMG20,21 by systematically predicting and validating drugs that target cell-state-specific tumorigenic regulators in vivo. ARACNe and VIPER were chosen over alternative network-based methods70,71,72,73,74,75,76, as they have been extensively benchmarked and repeatedly shown to nominate effective therapies12,14,16,17,57,77, including in clinical trials15,78. Here, we sought to apply their derivatives (OncoTarget/OncoTreat) to a biologically and clinically relevant question addressing tumor heterogeneity. Using this framework, we identified nine drugs predicted to invert cell-state-specific MR activity, with eight out of the nine demonstrating selective in vivo activity independent of underlying genetic alterations typically associated with drug sensitivity. Alternative network-based methods for assessing cell state dependencies could, in principle, be incorporated within this framework to extend its utility; however, this was not necessary given the strong and consistent in vivo validation. Direct in vivo validation was prioritized over comparative in silico or in vitro performance against alternative state-aware therapeutic selection approaches5,6,8,9,10, as it represents the most relevant benchmark for clinical translation.
Since in vitro DMG models are biased toward proliferative OPC-like states with idiosyncratic vulnerabilities, in vivo validation was essential, especially for minority states (AC and OC). The subcutaneous xenograft model reconstituted all DMG states, likely owing to 3D growth conditions and microenvironmental cues, allowing unbiased screening, free from BBB constraints, while a complementary syngeneic, orthotopic model confirmed drug efficacy in the native microenvironment context. Three OPC-targeting drugs—avapritinib, trametinib and dinaciclib—significantly improved survival. AC-targeting monotherapies had limited benefit, consistent with their minority cell state representation.
Critically, nearly all combinations targeting complementary cell states—including avapritinib/ruxolitinib, trametinib/ruxolitinib, dinaciclib/ruxolitinib and avapritinib/larotrectinib—outperformed monotherapies, with avapritinib/ruxolitinib producing the greatest survival benefit. These effects were not a result of cell-autonomous synergy but instead reflected effective co-depletion of distinct tumor cell states. This insight also suggests that combinations may remain effective under sequential dosing, reducing toxicity. For example, the synergy between avapritinib and larotrectinib suggests that triple therapy with avapritinib/larotrectinib/ruxolitinib (together targeting OPC, OC and AC states, respectively) could further overcome adaptive resistance driven by selection of minority states. Given that tumor progression is likely to occur whenever the dominant OPC states are not continuously targeted, alternating combinations (for example, avapritinib/ruxolitinib and avapritinib/larotrectinib) may offer a feasible strategy for sustained control.
To ensure translational relevance, we prioritized FDA-approved or late-stage compounds with known pediatric safety profiles. Although several drugs (for example, avapritinib, trametinib, larotrectinib) have shown limited monotherapy benefit in DMG32,79,80, our findings demonstrate that these agents may have a significant therapeutic impact when deployed in rational combinations targeting complementary states. Moreover, this work uncovered new vulnerabilities, including JAK1/3 dependency in AC-like cells. As previously described12,14,57,81, MR targeting can alter tumor state through diverse mechanisms—direct toxicity, differentiation or microenvironmental modulation—representing an important area for future investigation, potentially in combination with immunotherapies57.
Although promising, several limitations should be addressed in future work. First, even though this approach is generalizable, it requires perturbational profiles from models that preserve cell state MR activity, which may be challenging for some tumors. Short-term cultures from patient-derived tumors and advances in single-cell perturbation may help overcome this barrier. Conversely, using drug perturbational profiles offers a major advantage, driving the high in vivo validation rate, by enabling mechanistic prediction of both non-oncogene dependencies and their targeting drugs rather than relying on empirical in vitro viability screens. Second, although our study identified conserved regulatory modules across genetically diverse DMG tumors, in vivo validation was achieved in genetically similar models and modest cohort sizes; larger studies will be necessary to fully interrogate how genetic and epigenetic variation shapes response in DMG82. Third, optimal drug sequencing and dosing also require further study.
Children with DMG face a devastating diagnosis, with very limited treatment options. Altogether, this study identified non-oncogene drivers of DMG tumor cell states and promising drug combinations for clinical translation. Importantly, the study defined the DMG proteome-wide MoA for 372 clinically relevant drugs and validated OncoTarget/OncoTreat predictions, providing a platform to prioritize therapies for individual patients with DMG from bulk RNA-seq or to explore additional combinations. Furthermore, the underlying framework is agnostic to tumor type and can be adapted to identify effective combinations for any tumors with complex intratumor heterogeneity.
Rational prediction of effective drug combinations remains an elusive challenge in precision oncology. Our approach achieved 89% validation for predicted cell-state-specific agents and demonstrated that virtually all combinations targeting complementary states improved survival in vivo. A natural extension is to identify drugs that reprogram resistant states into sensitive ones. Together, these findings establish a robust, tumor-agnostic and mutation-agnostic framework for mechanism-based drug combination predictions, laying the groundwork for precision combination therapy across heterogeneous cancers.
Methods
Ethics and consent
No new human participants were prospectively enrolled. Publicly available human tumor RNA-seq datasets were analyzed under IRB protocol AAAS5252. All animal experiments were approved by the Columbia University Institute of Comparative Medicine and the Institutional Animal Care and Use Committee under protocol AC-AABL655, and adhered to institutional guidelines in full. Mice were maintained under barrier conditions and housed in ventilated racks with reverse-osmosis-purified water on a 12:12 h light–dark cycle (07:00–19:00 h) at 20–26°C and 30–70% humidity. Animals were killed if tumors exceeded ethical size limits (>10% total body weight or 2 cm in diameter), impaired posture or became necrotic or ulcerated. Maximal tumor size and burden were not exceeded in any experiment.
DMG patient scRNA-seq dataset and gene expression analysis
Publicly available scRNA-seq data from six DMG tumor biopsies (raw FASTQ files) were obtained from a previous publication20 and aligned to GRCh38 using Kallisto to derive single-cell count matrices. Gene expression data were processed in R using Seurat (v.4.3.0) with SCTransform normalization and scaling, followed by anchor-based integration (FindIntegrationAnchors, IntegrateData). Integrated data were projected onto the top 50 principal components (RunPCA) and visualized using uniform manifold approximation and projection (UMAP) (RunUMAP (Seurat v.4.3.0) with umap-learn, Pearson correlation). Unsupervised clustering was performed using the resolution-optimized Louvain algorithm (as implemented in Seurat). For regulatory network inference, metaCells were generated within each cluster by summing SCTransform-corrected counts from the ten nearest neighbors (Pearson distance), and metaCell expression profiles were re-integrated using Seurat’s anchor-based workflow.
DMG single-cell regulatory networks
Context-specific regulatory networks were inferred using ARACNe-AP (github.com/califano-lab/ARACNe-AP) to model bivariate regulatory interactions between predefined transcriptional regulators (github.com/califano-lab/tf_cotf_signalling_list) and candidate targets using standard parameters (zero data processing inequality tolerance and a mutual information P value threshold of 10−8 estimated by permutation). Cluster-specific networks were generated from normalized metaCell expression profiles to produce lineage-specific networks spanning tumor and non-tumor compartments. For tumor-specific analyses, patient-specific DMG networks were inferred independently using log2(counts per million (CPM) / 10 + 1)-normalized expression across malignant cells20. Patient-specific networks varied in size (714–2,469 regulators with ≥50 targets per network) and collectively enabled protein activity inference by metaVIPER (VIPER Bioconductor package, v.1.34.0) for 2,821 regulatory proteins.
DMG single-cell protein activity inference
Single-cell protein activity was inferred from SCTransform-scaled, anchor-integrated metaCell gene expression profiles using metaVIPER37, which integrates multiple regulatory networks on a protein-by-protein basis using squared normalized enrichment scores (NES). Protein activity was computed for all cells using cluster-based single-cell ARACNe networks and for tumor-only cells using the six patient-specific tumor networks. Protein activity matrices were loaded into a Seurat Object, projected onto the top 50 principal components and visualized by UMAP as before.
Resolution-optimized Louvain clustering
Clustering of cells by gene expression and protein activity, for both all-cell and tumor-only analyses, was performed using a resolution-optimized Louvain algorithm41 implemented with Seurat’s FindNeighbors and FindClusters functions, with objective selection of the resolution parameter by maximizing average silhouette score. Silhouette scores were computed across resolution values from 0.01 to 1.0 (step size, 0.01) using 100 random subsamples of 1,000 cells to estimate the mean silhouette score and variance (Supplementary Fig. 1f).
Identifying and removing non-malignant cells
Non-malignant immune cells were identified by enrichment of a microglia gene signature (SALL1, HEXB, FCRLS, GPR43, CX3CR1, TMEM119, TREM2, P2RY12, MERTK, PROS1, SIGLECH85) assessed by GSEA (atools gsea; 100 permutations) in both gene expression and protein activity profiles. Although the microglia signature and CD33 were not detectable by gene expression, VIPER-inferred protein activity robustly annotated immune clusters (Supplementary Fig. 1a–d).
Copy number variation (CNV) was inferred from gene expression using InferCNV (v.1.20.0) with immune clusters as the reference. Tumor clusters exhibited copy number alterations in at least a subset of cells (Supplementary Fig. 1e). Given inter-patient variability in CNV burden and the presence of DMGs with low CNV, malignant identity was assigned at the cluster level rather than to individual cells, and non-malignant clusters were excluded from tumor-only analyses.
Single-cell pathway enrichment
Differential gene expression between protein activity-defined clusters in 3,032 tumor cells was assessed using the Wilcoxon test, comparing each cluster to all others (Supplementary Table 1). Significantly upregulated genes (Bonferroni P ≤ 0.05; positive log2(fold change)) were used as cluster-specific gene sets. Pathway enrichment was performed by overrepresentation analysis using enrichR (v.3.2) with the ‘MSigDB Hallmark 2020’ library, with FDR correction across pathways (FDR ≤ 0.05 considered significant).
DMG single-cell validation dataset analysis
To validate DMG cell states and associated MRs, we analyzed an independent single-cell dataset generated from fresh single cells from pediatric patients (age <19 years; eight samples, 1,629 high-quality tumor cells)21. Post-filtered gene expression data were obtained from GEO (GSE184357) and processed identically to the discovery cohort, including protein activity inference by metaVIPER using the prior DMG single-cell regulatory networks and resolution-optimized Louvain clustering. Cell state assignment was determined by one-sided GSEA (100 permutations) assessing enrichment of the top 50 cell state MRs from the discovery cohort in single-cell protein activity profiles of the validation dataset; cells were labeled by the most significantly enriched state. Concordance between unsupervised clusters and supervised state annotations was assessed by χ2 analysis. MR conservation between cohorts was assessed by aREA (viperSimilarity function, VIPER package, v.1.34.0), comparing integrated cell state protein activity signatures.
An integrated analysis of all 14 DMG samples (4,661 tumor cells), processed identically to the discovery cohort, was performed to validate protein activity-based cell states and confirm their identity by lineage marker enrichment. Conservation of cell state MRs across individual samples and between protein activity signatures generated using single-cell versus bulk-derived DMG networks was assessed using viperSimilarity on this integrated dataset.
DMG bulk RNA-seq datasets
Publicly available bulk RNA-seq data and clinical annotations from 122 patients with DMG were obtained from three cohorts (Supplementary Table 2): two from the Pediatric Brain Tumor Atlas (PBTA) PBTA:PNOC86 (n = 31) and PBTA:CBTTC87 (n = 47) through the Gabriella Miller Kids First Data Resource Center, and one from St. Jude (n = 44) from a previous publication26. Samples were selected based on diagnostic labels and review of clinical annotations with a pediatric neuro-oncologist to include high-grade gliomas arising in midline structures (brainstem, thalamus, spinal cord) with molecular features consistent with DMG. To minimize batch effects, raw FASTQ or BAM files (converted using Picard SamToFastq, v.2.25.0) were uniformly processed. Gene expression was quantified with Kallisto using GRCh38 cDNA and ncRNA references and normalized as transcripts per million.
DMG bulk regulatory network
Given that some regulatory proteins lack sufficient dynamic range in single-cell profiles, we generated a lineage-related regulatory network from DMG bulk RNA-seq data. Given the limited sample size, networks were inferred using ARACNe3 (github.com/califano-lab/ARACNe3), which uses a maximum-entropy-based analytical null model to reduce sample and bootstrap requirements19,88. Putative regulators were defined as in the single-cell networks. To minimize batch effects and autopsy–biopsy bias, a consensus network was constructed from ARACNe3 networks inferred separately within five subsets stratified by cohort and specimen type (CBTTC_autopsy, n = 15; CBTTC_biopsy, n = 30; PNOC_biopsy, n = 31; StJude_autopsy, n = 28; StJude_biopsy, n = 16; 120 total profiles). Individual networks were pruned using a mutual information threshold controlling FDR at 0.50, followed by MaxEnt (data processing inequality) pruning89. Regulons were integrated by consensus scoring with association weight and association mode averaged across networks weighted by subset size. The final network comprised regulons with ≥50 targets for 6,138 regulatory proteins.
Single-cell and bulk-derived networks yielded highly congruent cell state MRs (Extended Data Fig. 1h). The bulk network was used for analyses requiring broader regulator coverage; specifically, synthetic bulk protein activity analyses to assess enrichment of CRISPR-essential genes and for bulk OncoTarget/OncoTreat (via n1platform, v.1.3.23). Single-cell networks were used for all other analyses.
Culture of DMG cells
Previously extensively validated DMG cell lines were sourced from collaborators, including SF8628 RRID: CVCL_IT46 (female) and SF7761 RRID: CVCL_IT45 (female), gifted by R. Hashizume; SU-DIPG-IV RRID: CVCL_IT39 (female), SU-DIPG-VI RRID: CVCL_IT40 (female), SU-DIPG-XVII RRID: CVCL_C1MW (male) and SU-DIPG-XXXVI RRID: CVCL_C1N5 (female), gifted by M. Monje; and DIPG4423, gifted by O. Becher. SU-DIPG-IV (DIPG4), SU-DIPG-VI (DIPG6), SU-DIPG-XVII (DIPG17), SU-DIPG-XXXVI (DIPG36) and SF7761 cells were cultured in Knockout DMEM/F-12 (Gibco, Fisher Scientific, 12-660-012), Stempro Neural Supplement (Gibco, Fisher Scientific, A1050801), EGF Recombinant Human Protein (Gibco, Fisher Scientific, PHG0313), Recombinant Human FGF-Basic (aa 10–155; Gibco, Fisher Scientific, PHG0021), 1% penicillin–streptomycin solution (Sigma-Aldrich, P4458), and 1% L-glutamine (Corning, Fisher Scientific, MT25005Cl). SF8628 cells were cultured in DMEM, 10% FBS (Sigma-Aldrich, F2442), 1% penicillin–streptomycin solution and 1% L-glutamine. DIPG4423 cells were cultured in Mouse Neurocult media (Stemcell Technologies, 05700), Mouse and Rat Proliferation Supplement 10% (Stemcell Technologies, 05701), Recombinant Human FGF (20 ng ml−1; PeproTech, 100-18B), Recombinant Human EGF (10 ng ml−1; PeproTech, AF-100-15), Heparin (2 μg ml−1; Stemcell Technologies) and 1% penicilin–streptomycin solution. All cell lines tested negative for mycoplasma contamination. Cell lines were confirmed to be transcriptionally consistent with DMG patient profiles by bulk and scRNA-seq.
Generation of Cas9-expressing DMG cells
Lentivirus was produced in HEK293T cells maintained in DMEM (Gibco, 11995073) with 10% FBS (Sigma-Aldrich, F2442). Cells were transfected using 18 μl of 2 mg ml−1 polyethylenimine (PEI 25000; Polysciences, 23966-1) with 6 μg plasmid DNA, 4.5 μg psPAX2 (Addgene, 12260) and 1.5 μg pMD2.G (Addgene, 12259) mixed in Opti-MEM (Gibco, 31985062) per 10 cm dish. Media was replaced 12 h post transfection, and viral supernatant was collected at 48 h and 72 h, filtered (0.45 µm), precipitated with PEG-6000 (Santa Cruz Biotechnology, sc-302016), resuspended and pooled. For the transcription factor single guide RNA (sgRNA) library, lentivirus was produced at a scale maintaining >1,000 representation.
To generate Cas9-expressing cells, DMG cells (DIPG4, DIPG17, SF8628) were spin-infected with LentiV_Cas9_puro lentivirus (gift from C. R. Vakoc to Z. Zhang) at a multiplicity of infection of ~1.5 (1,200 rpm, 25°C, 1 h). The media was refreshed 24 h post infection, and cells were selected with Puromycin (1 μg ml−1, 3 days; Invivogen, ant-pr), followed by a second infection and selection with Puromycin (1.5 μg ml−1, 3 days). Cas9 expression was confirmed by western blot using anti-Cas9 antibody (Cell Signaling Technology, 65832), and cutting efficiency was validated using sgRNA targeting CDK1 and PCNA.
Generation of luciferase-expressing cells
DMG cells (DIPG4, DIPG17, SF8628) were spin-infected with firefly-LUC_pHR-SIN-CSGW lentivirus (gift from R. Hashizume to Z. Zhang) at a multiplicity of infection of ~2.5 (1,200 rpm, 25°C, 1 h). The media was refreshed 24 h post infection. Luciferase expression was confirmed by immunofluorescence using anti-Luciferase antibody (Abcam, ab21176), and cell populations with >90% luciferase-positive cells were used for experiments.
sgRNA library generation and CRISPR–Cas9 pooled screening
A transcription factor domain-focused sgRNA library (gift from C. R. Vakoc to Z. Zhang) was amplified in ElectroMAX Stbl4 Competent Cells (Invitrogen, 11635018) according to the manufacturer’s instructions. Cas9-expressing DMG cells were spin-infected with the sgRNA lentiviral library and maintained at >1,000 representation throughout the screen. Samples were collected at day 3 post infection (T0) and after 12 population doublings (T1). Genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, 69504), and barcoded sequencing libraries were prepared72, pooled and sequenced by paired-end NextSeq (Illumina) for each of the three DMG cell lines. The sgRNA library contained approximately six sgRNAs per gene targeting 1,486 genes, including 1,373 transcription factors, 62 core-essential positive controls and 51 non-targeting negative controls (Supplementary Table 3).
Analysis of CRISPR–Cas9 pooled knockout screens
CRISPR screens were analyzed using MAGeCK10 with Robust Rank Aggregation and total read count normalization (default settings)10. Replicates were analyzed independently (T1 vs T0) to compute log(fold change) essentiality signatures for each cell line. Core-essential genes (DepMap12 reference) were excluded, and results were integrated across cell lines by median fold change. Separation between core-essential and non-targeting control sgRNAs was validated by Kolmogorov–Smirnov testing of their empirical cumulative distribution functions (Supplementary Fig. 2a). All raw data and results are reported in Supplementary Table 3.
Enrichment of CRISPR-essential genes in DMG MRs
Using the DMG bulk regulatory network, VIPER protein activity was inferred for tumorigenic gene expression signatures defined relative to the centroid of 246 GTEx normal caudate samples (per-gene z-score transformation of log2(CPM + 1)-normalized expression). Protein activity was computed for synthetic bulk profiles generated by aggregating reads from all (1) malignant single cells, (2) each of the seven DMG cell states and (3) malignant cells from each individual patient.
Tumorigenic MRs (that is, top and bottom 50 regulators per signature) were tested for enrichment in DMG CRISPR-essential genes, defined either from the integrated essentiality signature or from individual cell lines (−log2(fold change), FDR ≤ 0.05). Enrichment was assessed by one-sided GSEA, with significance estimated from 200 permutations and FDR correction. The union of genes in leading edges across analyses was designated DMG-essential MRs. The same approach was applied to assess enrichment of VIPER-inferred tumorigenic MRs in cell-line-specific CRISPR-essential gene sets from six DMG cell lines profiled by bulk RNA-seq.
DMG cell line model fidelity by OncoMatch
Bulk RNA-seq data (FASTQ files) from six DMG cell lines (DIPG4, DIPG6, DIPG17, DIPG36, SF7761, SF8628) were quantified by Kallisto. Protein activity was inferred by VIPER relative to the GTEx caudate centroid per the prior section. Model fidelity was evaluated by MR conservation using aREA within OncoMatch17, testing enrichment of the top and bottom 50 patient MRs in cell line protein activity signatures (FDR ≤ 10−5 was considered significant). SF8628 and DIPG6 were selected as complementary high-fidelity models.
Drug perturbation profiling by PLATE-seq
A library of 372 FDA-approved and late-stage experimental oncology compounds was profiled in SF8628 and DIPG6 cells (Supplementary Table 4). To determine sub-lethal dosing (48 h EC20), cells were seeded in 384-well plates (Greiner 781080; columns 1–24, 50 μl media) at 1,000 cells per well (DIPG6) or 500 cells per well (SF8628), incubated at room temperature (20–25 °C) for 30 min then overnight in an incubator (37 °C, 5% CO2) for ~12 h, after which compounds were dispensed in ten doses by an Echo 550 acoustic dispenser after removal of 40 μl of media per well (plates done in triplicates). Cell viability was measured after 48 h by the addition of 25 μl of CellTiter-Glo (Promega Corp, G7570) per well with enhanced luminescence readout (EnVision, PerkinElmer) after shaking the plates for 5 min. Values were normalized to plate-matched dimethylsulfoxide (DMSO) controls, and EC20 values were computed using Pipeline Pilot (Dassault Systèmes).
Drug perturbation profiling was performed using the lower of EC20 or Cmax (≤10 μM) to avoid stress-related transcriptional responses16,89,90. For each cell line, two biological replicates were profiled, with each plate containing 372 drug treatments, six DMSO controls and six untreated samples. After 24 h, plates were spun down at 295g for 1 min. Media was removed, and cells were resuspended in 30 μl of Turbo Capture Lysis buffer (Qiagen, 1070498) containing 1% beta-mercaptoethanol. Multiplexed, low-depth RNA-seq libraries (less than five million reads per sample) were generated using the PLATE-seq microfluidic automation platform56 at the Columbia JP Sulzberger Genome Center High-Throughput Screening facility.
Reads were quantified using Kallisto with the GRCh38 reference and demultiplexed by the barcoded reads into individual drug counts. Expression was normalized by equi-variance transformation (DESeq), cross-plate batch effects were corrected with ComBat (sva package) and differential expression relative to plate-matched DMSO controls was modeled using limma (v.3.56.2). Drug-induced differential protein activity was inferred by metaVIPER from the moderated t-statistics using the DMG single-cell networks.
Single-cell OncoTarget and OncoTreat
Single-cell protein activity across tumor cells was inferred by metaVIPER from tumorigenic gene expression signatures (per-gene z-score transformation of log10(CPM + 1)-normalized metaCell expression relative to GTEx caudate centroid) using DMG single-cell tumor regulatory networks. Within each state, single-cell NES were integrated by Stouffer’s method to generate state-level protein activity signatures.
OncoTarget was performed by restricting state-level signatures to 180 curated actionable regulatory proteins (targets of high-affinity inhibitors)17. OncoTreat assessed each drug’s ability to invert the top or bottom 50 tumorigenic MRs per state based on enrichment (aREA) of tumorigenic MRs within drug-induced protein activity signatures from DIPG6 and SF8628 (Supplementary Table 4). NES across cell lines were integrated by Stouffer’s method, weighted by OncoMatch fidelity scores. Significance was computed using a one-sided pnorm with Bonferroni correction across proteins (OncoTarget) or drugs (OncoTreat) within each state (Bonferroni P ≤ 10−5 considered significant; Supplementary Table 4).
Single-cell isolation and gel beads-in-emulsion generation in DMG cell lines
SF8628 cells were detached using room-temperature ACCUTASE (Sigma-Aldrich, A6964). Neurospheres were collected, centrifuged (250g, 6 min, 4 °C), resuspended in ACCUTASE (1 ml, 3 min) and mechanically dissociated by pipetting (~20 times). Cells were filtered through a 70 μm strainer, washed three times in scRNA-seq buffer (HBBS + 0.04% BSA; Fisher Scientific, 14-175-095; Miltenyi Biotec, 130-091-376) and assessed for viability and concentrations using a Countess II. Cells were resuspended in 1 × 106 cells per ml and processed for single-cell gene expression capture using the Chromium 3′ Library and Gel Bead Kit (10× Genomics). Libraries were sequenced on an Illumina NovaSeq 6000 at the Columbia University Single Cell Analysis Core.
Drug dilutions
All drugs were received as powders and prepared using a common vehicle consisting of 30% (15 ml) polyethylene glycol 400 (PEG 400; Selleck Chemicals, S6705), 5% (2.5 ml) polyoxyethylene sorbitan monooleate (Tween 80; Selleck Chemicals, S6702) and 65% (32.5 ml) 5% dextrose in water (D5W; Fisher Scientific, NC0215480) (PTD). Vehicle volumes were prepared based on cohort size and dosing duration (ten total dosing days, Monday–Friday). Drug doses (mg kg−1) were determined per Institutional Animal Care and Use Committee protocol AC-AABL6551 (Y2 M12), assuming a 30 g average mouse weight and a dosing volume of 10 µl g−1. After calculating drug mass and volume, 200 µl of N-methyl pyrrolidinone (Sigma-Aldrich, M79603) was added to enhance solubility, and the remaining volume was filled with PTD. Solutions were vortexed or probe-sonicated as needed to ensure homogeneity.
Subcutaneous DIPG17 xenograft experiments
Juvenile 6-week-old male and female (1:1 ratio) NOD.Cg-Prkdcscid/J (Envigo) mice were inoculated subcutaneously (left flanks) with 500,000 DIPG17-luciferase cells suspended 1:1 in complete medium and Matrigel (200 μl total; Fisher Scientific, CB-40234). Tumor growth was monitored starting 2 weeks post injection using the IVIS Spectrum Optical Imaging System (PerkinElmer). Mice received intraperitoneal luciferin (15 mg ml−1, 10 μl g−1), were anesthetized with isoflurane (2 l min−1) and imaged 10 min post injection.
Once tumors reached ~100 mm3 (range, 75–185 mm3), mice were randomized to vehicle or treatment arms and treated for 5 days: vehicle (n = 4), larotrectinib 200 mg kg−1 per dose oral gavage (PO) every day (QD) (n = 2), trametinib 1 mg kg−1 per dose PO QD (n = 2), dinaciclib 40 mg kg−1 per dose intraperitoneally (Monday, Wednesday, Friday; n = 2), avapritinib 30 mg kg−1 per dose PO QD (n = 3), ruxolitinib 90 mg kg−1 per dose PO twice per day (n = 2), venetoclax 100 mg kg−1 per dose PO QD (n = 3), etoposide 8 mg kg−1 per dose intraperitoneally QD (n = 3), mocetinostat 50 mg kg−1 per dose PO QD (n = 2) and napabucasin 10 mg kg−1 per dose (Monday, Wednesday, Friday; n = 3). All drugs were obtained from Selleckchem. Mice were killed 2 h after the final dose on day 5 for tumor isolation and scRNA-seq.
High-frequency ultrasound imaging system for tumor volume assessment in subcutaneous xenografts
Three-dimensional ultrasound imaging was obtained for each xenograft using the VEVO 3100 high-frequency micro-ultrasound system (VisualSonics). Mice were anesthetized with 1.5–2% isoflurane in oxygen (0.75 l min−1) and immobilized for imaging. Xenografts were coated with warmed (37 °C) Aquasonic 100 US gel (Parker Laboratories), centered in the imaging plane and imaged across the full xenograft length. Images were imported into VEVO software for tumor volume calculation. The percent volume change was calculated as:
Differences in tumor volume change between each treatment arm and vehicle control were assessed by Welch’s t-test (P ≤ 0.05 considered significant).
ScRNA-seq of DIPG17 subcutaneous xenografts
Following tumor isolation, fat, fibrotic and necrotic tissues were removed, and tumors were cut into 1–2 mm pieces. Tissue was transferred to gentleMACS C Tubes (Miltenyi Biotec, 130-093-237) containing Knockout DMEM/F-12 (4.7 ml; Gibco, 12-660-012), Enzyme H (200 μl), Enzyme R (100 μl) and Enzyme A (25 μl) from the Human Tumor Dissociation Kit (Miltenyi Biotec, 130-095-929). Samples were dissociated using the gentleMACS Dissociator (‘Medium’ tumor program; 37C_h_TDK_2) for 18 min, filtered through a 70 μm strainer and centrifuged (250g, 8 min, 4 °C). Pellets were resuspended in RBC lysis buffer (400 μl; Miltenyi Biotec, 130-094-183), incubated on ice for 2 min, quenched with ≥3× buffer volume, centrifuged (250g, 5 min) and resuspended in HBBS + 0.04% BSA. Samples were assessed for viability and concentration using the Countess II and submitted to the Columbia University Single Cell Analysis Core, where 3′ scRNA-seq libraries (10× Genomics) were prepared and sequenced on an Illumina NovaSeq 6000. Raw data were processed using Cell Ranger (default parameters) to generate gene-cell count matrices.
Analysis of drug-perturbed scRNA-seq in DIPG17 subcutaneous xenograft tumors
Low-quality cells and genes without reads were removed, including cells with <1,000 or >100,000 total unique molecular identifiers (UMIs) or mitochondrial transcript fraction >15%. Filtered data were normalized (SCTransform) and gene expression signatures generated by scaling SCTransform-normalized counts relative to all vehicle-treated tumor cells.
Human versus mouse origin of profiled cells was determined by consensus alignment of RNA-seq reads to the human and mouse reference genomes (Supplementary Fig. 3c). The distribution of human-aligned UMIs across cells was modeled using a two-component finite mixture model, with parameters estimated by expectation-maximization (em function)91,92, to distinguish human tumor cells from mouse-infiltrated cells. The fitted model identified a bimodal distribution corresponding to mouse-derived (low human UMI fraction) and human-derived (high human UMI fraction) populations. A cutoff separating the two models was derived from the fitted model (cutoff function), yielding a negligible probability of misclassification for cells exceeding this threshold (cutoff, 43.32%; Supplementary Fig. 3c). Based on this analysis, cells with >50% human-aligned UMIs were retained as human tumor cells for downstream analyses.
Protein activity (NES) was inferred from vehicle-scaled gene expression signatures for all human tumor cells using metaVIPER and DMG patient single-cell regulatory networks for 2,821 regulatory proteins across 95,687 tumor cells spanning all treatment conditions.
Assessing in vitro and in vivo DMG model fidelity
To assign cell identities to the 11,325 cell line and 95,687 subcutaneous tumor single cells, we performed GSEA evaluating enrichment of the top 50 activated DMG patient cell state MRs (Supplementary Table 1) within model single-cell protein activity signatures, with NES estimated from 100 permutations. Significance was computed as 1 – pnorm(NES), with FDR correction across states. Based on MR similarity across related states, cells were assigned to OPC (OPCQ or OPC), OPCC, OC (OC or OPC/OC), AC or OPC/AC identities according to their most significantly enriched state (FDR ≤ 0.05 considered significant).
To evaluate conservation of cell-state-specific OncoTarget/OncoTreat predictions, synthetic bulk differential protein activity signatures were generated by metaVIPER from cells assigned to each state, computed relative to the differentiated OC state within the same model (given the absence of an appropriate normal reference for subcutaneous DMG tumors). OncoTarget/OncoTreat was then performed as described for tumors from patients with DMG.
Evaluation of drug-induced cell state depletion
For each treatment arm, drug-induced changes in cell state composition were quantified as the log(fold change) in the proportion of cells assigned to each state in drug-treated versus vehicle-treated tumors. Biological replicates were analyzed independently, and mean log(fold changes) with standard errors were calculated across replicates. Given shared drug predictions across related states, results were summarized into three composite groups: OPC (OPCQ, OPC, OPCC), OC (OC, OPC/OC) and AC (AC, OPC/AC).
For each treatment arm, bar plots were generated showing the mean and s.e.m. (s.d/√n) of cell state proportions in drug-treated (n = 2–3) versus vehicle-treated (n = 4) samples (Supplementary Fig. 4). Differences in cell state distributions between drug-treated and vehicle-treated tumors were assessed using χ2 tests (each state versus all others), with FDR correction across states and treatment arms (Supplementary Table 5).
Pharmacodynamic assessment of MR activity reversal in vivo
Differential protein activity signatures were inferred by metaVIPER for each cell state by comparing drug-treated and vehicle-treated cells using DMG single-cell regulatory networks. Within each treatment arm, state-specific signatures were integrated across replicates by Stouffer’s method to generate in vivo drug-versus-vehicle profiles. MR inversion was quantified by two-sided aREA enrichment of the top and bottom 50 human DMG tumorigenic MRs within each respective state-specific signature. For OncoTarget-predicted drugs, changes in VIPER-inferred activity of the targeted MR additionally evaluated on-target drug activity.
Generation of a syngeneic orthotopic DMG model
DIPG4423 murine cells were detached using ACCUTASE, centrifuged (250g, 6 min, 4 °C), resuspended in ACCUTASE (1 ml, 3 min), pipetted 20 times, washed with fresh media (3 ml), centrifuged again (250g, 6 min), filtered through a 70 μm strainer and resuspended at 100,000 cells per μl. Cell viability was confirmed using a Countess 3FL Automated Cell Counter.
Under sterile conditions, 6-week-old male B6(Cg)-Tyrc-2J/J mice (Jackson Laboratory) were anesthetized with 1–2% isoflurane in oxygen (0.75 l min−1) and immobilized in a stereotactic instrument (Stoelting, 51730). Only male mice were used because of known significant differences in intracranial tumor growth between male and female mice to avoid confounding (male mice exhibit faster growth rates). A 1 cm midline scalp incision was made, and a 1 mm burr hole was drilled 1.5 mm posterior to the lambda and 1.5 mm lateral to the sagittal suture. DIPG4423 cells (100,000 cells in 1 μl) were injected 5 mm below the skull surface at 0.1 ml min−1 over 10 min using a Hamilton syringe (Hamilton Company, 830314). The syringe was slowly removed 1 mm every 30 s, the burr hole covered with bone wax (Fisher Scientific, 50-118-0260) and the incision closed with sutures (Fisher Scientific, 50-118-0846). Triple antibiotic ointment was applied to the wound. Postoperative analgesia (buprenorphine, 0.05 mg kg−1 intraperitoneally) was administered (Covetrus, approved by DOH/DEA), mice recovered on a heating pad and the wound was monitored daily for 7 days. Tumor presence and location were confirmed by MRI 12 days post implantation.
Single-cell dissociation and sequencing of orthotopic models
Untreated tumor-bearing mice were killed 3 weeks post implantation for tumor single-cell dissociation, along with one normal brainstem control. Mice were anesthetized with 2% isofluorane and perfused with cold PBS (8 min and/or liver turned pale). Brainstems were dissected and placed in isolation media (RPMI 1640 with GlutaMAX and HEPES (Gibco, 72-400-120) with 0.01% 50X B27 supplement (Gibco, 17-504-044)). Tissue was homogenized using a glass tissue grinder, filtered through a 70 μm filter and centrifuged (300g, 10 min, 4 °C). Pellets were resuspended in isolation media (1 ml) and incubated with myelin removal beads (100 μl, 15 min; Miltenyi Biotec, 130-096-433), followed by depletion using LS-positive separation columns (Miltenyi Biotec, 130-042-401). After washing in single-cell buffer (HBBS + 0.04% BSA), cells were resuspended in RBC lysis buffer (400 μl, 2 min on ice), followed by four additional washes in single-cell buffer (1 ml), centrifugation (250g, 5 min) and resuspension at ~1 × 106 cells per ml. Cell counts and viability were assessed using a Countess 3FL Automated Cell Counter, and samples were submitted to the Columbia University Single Cell Analysis Core for 3′ scRNA-seq (10× Genomics), processed as for subcutaneous tumors. This yielded 20,362 tumor-derived and 2,978 normal brainstem high-quality single cells.
Assessing orthotopic model fidelity
Across two tumor-derived and one normal brainstem (wild-type) sample, unsupervised clustering of gene expression profiles using the standard Seurat pipeline with singleR annotation (v.2.2.0, mouse.rnaseq reference) identified glial, neuronal and immune populations, including microglia. Immune clusters were excluded before re-clustering putative tumor cells. Malignant clusters from the two tumor samples were confirmed by inferCNV analysis using wild-type cells as a reference (Extended Data Fig. 6c,d), yielding 8,013 malignant cells. Tumor cell expression data were normalized and scaled using SCTransform. Given species-specific regulatory interactions, murine DMG single-cell regulatory networks were independently inferred from each tumor sample using ARACNe-AP, as described before, and interrogated by metaVIPER to infer protein activity. Protein activity profiles were clustered using resolution-optimized Louvain clustering, and cell state identities were assigned by enrichment of human DMG cell state MRs assessed by GSEA (100 permutations), as for subcutaneous tumors. MR conservation between orthotopic and patient-derived cell states was confirmed by aREA analysis using viperSimilarity. Conservation of cell-state-specific OncoTarget/OncoTreat predictions was further assessed using synthetic bulk protein activity profiles computed relative to the GTEx caudate centroid, as performed for tumors from patients with DMG.
MRI and analysis
Tumor location, growth and volume were assessed using a 9.4T MRI system (Bruker Medical) with T2-weighted TurboRARE sequences. Mice were anesthetized with 1–2% isoflurane in oxygen (0.75 l min−1) and imaged using a 35 mm birdcage coil. DICOM images were analyzed in 3D Slicer (www.slicer.org) to calculate tumor volumes.
Histopathology
Mice were killed 3 weeks post injection (vehicle or untreated) or upon development of neurological symptoms (treated). Whole brains were fixed in formaldehyde (15 ml) overnight, sagittally sectioned and further fixed at room temperature. After 12 h, the tissue was transferred to cold PBS and stored at 4 °C overnight. Samples were then processed for paraffin embedding, H&E staining and whole-slide scanning by the Columbia University Irving Medical Center Histology Core. Whole-slide images (.svs) were analyzed using Orbit Image Analysis Software (v.3.64).
Syngeneic orthotopic survival studies
At 2 weeks post injection, following MRI confirmation of tumor establishment, mice were randomized to monotherapy or combination treatment arms. Given the number of treatment arms, including individual drugs and drug combinations, cohort sizes were selected to ensure a minimum of three independent biological replicates per treatment arm, consistent with exploratory in vivo efficacy studies. The vehicle-treated control arm included nine mice to establish a robust baseline distribution. Across experiments, the median cohort size was four mice per monotherapy arm and 3.5 mice per combination therapy arm (range, 3–5).
Drug dosing and routes matched those used in subcutaneous xenograft studies. All drugs were administered on a 5 days on, 2 days off schedule. Combination therapies paired agents targeting distinct cell states and included avapritinib with larotrectinib, venetoclax or ruxolitinib; trametinib with venetoclax or ruxolitinib; and dinaciclib with venetoclax or ruxolitinib. Toxicity was monitored by weight, temperature and behavior. The dinaciclib/venetoclax arm was discontinued for toxicity (rapid weight loss). Drugs were staggered by ≥6 h when possible, or by 3 h when combined with twice-per-day ruxolitinib.
Mice were killed upon development of significant neurological symptoms or >15% weight loss. Survival curves were generated using GraphPad Prism (v.9.5.1) and compared using the log-rank (Mantel–Cox) test (P ≤ 0.05 considered significant).
Synergy studies
Dose–response curves were generated by plating DIPG36 and DIPG4423 cells (1,000 cells per well), or SF8628 (500 cells per well) in 384-well white tissue culture plates (Fisher Scientific, Greiner Bio-One, 07-000-074) and incubating for 24 h at 37 °C with 5% CO2. Compounds were dispensed using an HP Digital Dispenser and titrated from 40 mM to 2.6 nM in triplicate (0.4% DMSO). After 48 h, CellTiter-Glo (25 μl; Promega Corp, G7570) was added and luminescence measured on an EnVision Multi-Label plate reader (Revvity). Viability was normalized to on-plate DMSO and thimerosal controls using Pipeline Pilot.
For combination studies, cells were plated as above and treated after 24 h with drug pairs dispensed in 10 × 10 matrices, using the previously determined IC50 as the maximum concentration. Matrices were run in triplicate with randomized placement and DMSO and thimerosal controls. After 48 h, viability was assessed as above. BLISS synergy scores and plots were generated using SynergyFinder 3.0 (synergyfinder.fimm.fi)68.
Functional validation of bulk OncoTarget/OncoTreat predictions
Bulk RNA-seq counts and matched in vitro drug screening data from three DMG tumors (MTB36, MTB37, MTB41) were obtained through a functional precision medicine program at Rady Children’s Hospital69. For each gene expression sample, log2(CPM + 1)-normalized counts were scaled to the GTEx caudate centroid, and differential protein activity was inferred by VIPER using the DMG bulk network. OncoTarget/OncoTreat analyses were performed as previously described. Predicted drugs were compared with functional screening results by classifying compounds as effective (IC50 ≤ 10 μM and cell viability of ≤50%) or inactive. Only compounds reducing viability at ≤10 μM were considered, as higher concentrations were not profiled by PLATE-seq. Predictive performance was evaluated using odds ratios, specificity, positive predictive value and overall accuracy, and the association between OncoTreat NES and drug potency (log10(IC50)) was assessed using two-sided Spearman rank correlation.
Statistics and reproducibility
No statistical method was used to predetermine sample size. For computational analyses, sample sizes were determined by the availability of public datasets and predefined inclusion criteria. For animal studies, mice were randomly assigned to treatment arms after tumors reached target volumes; experiments included at least two biological replicates for single-cell analyses and at least three biological replicates for tumor volume and survival studies. Unless otherwise stated, no data were excluded except for predefined quality-control filtering for single-cell data. Where applicable, experiments included technical and/or biological replicates as detailed in the corresponding Methods subsections, and patient-derived single-cell findings were validated in an independent dataset. Investigators were not blinded to allocation during experiments or outcome assessment. Statistical analyses were performed in R (v.4.2.2) unless otherwise specified. Specific statistical tests and multiple-hypothesis corrections are described in the relevant Methods subsections.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The following source data generated as part of this work have been made available through the Gene Expression Omnibus (GEO) https://www.ncbi.nlm.nih.gov/geo: Drug screen RNA-seq data from 372 oncology compounds in two patient-derived DMG cell lines (PLATE-seq) (GSE264130), pooled CRISPR–Cas9 screen targeting all transcription factors in three DMG patient-derived cell lines (GSE267415), drug screen RNA-seq data on three DMG patient biopsies (GSE264412), scRNA-seq data of DIPG4423 syngeneic orthotopic brainstem tumors and normal brainstem (GSE262774) and scRNA-seq data of DIPG17 subcutaneous murine models (drug-treated and vehicle-treated) (GSE262775). Previously published DMG patient scRNA-seq data analyzed in this study are available under accession codes GSE102130 (ref. 20; raw FASTQ files were provided by the authors) and GSE184357 (ref. 21). Additionally, publicly available data were curated for 122 bulk RNA-seq profiles from three DMG datasets, including St. Jude26 (European Genome-phenome Archive (EGA) EGA0000100192), PNOC86 and CBTTC88). The latter two were downloaded from the Pediatric Brain Tumor Atlas (PBTA) through the Gabriella Miller Kids First Data Resource Center (kidsfirstdrc.org) (see Methods for details on criteria used for sample inclusion). The ARACNe regulatory networks used to run VIPER, including the DMG patient tumor single-cell networks, the DMG patient bulk network and the DIPG4423 syngeneic orthotopic mouse DMG tumor single-cell networks, are available on Figshare (https://doi.org/10.6084/m9.figshare.30002785; https://doi.org/10.6084/m9.figshare.29988670; https://doi.org/10.6084/m9.figshare.29988511 and https://doi.org/10.6084/m9.figshare.29988508). The same Figshare link additionally contains the VIPER-inferred regulatory protein activity NES matrix for all significantly activated proteins across the DMG patient tumor single cells from which cell states and their MRs were derived. Source data are provided with this paper.
Code availability
No novel software or standalone custom code packages were generated for this study. All computational analyses were performed using established, publicly available algorithms and software, with software versions, key parameters, statistical thresholds and analytical choices fully specified in the Methods and Reporting Summary; accordingly, all components, including source data, required to reproduce the analyses are publicly accessible. Regulatory network inference and protein activity analyses were performed using the ARACNe and VIPER algorithms, including their single-cell extensions, with source code, documentation and example workflows publicly available at the following repositories: https://doi.org/10.5281/zenodo.18665401; https://doi.org/10.5281/zenodo.18665521 and https://doi.org/10.5281/zenodo.18665411. Reasonable requests for clarification, scripts or configuration details will be fulfilled by the corresponding author (A.C.).
References
Alizadeh, A. A. et al. Toward understanding and exploiting tumor heterogeneity. Nat. Med. 21, 846–853 (2015).
McGranahan, N. & Swanton, C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168, 613–628 (2017).
Marusyk, A., Janiszewska, M. & Polyak, K. Intratumor heterogeneity: the Rosetta Stone of therapy resistance. Cancer Cell 37, 471–484 (2020).
Dagogo-Jack, I. & Shaw, A. T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81–94 (2018).
Fustero-Torre, C. et al. Beyondcell: targeting cancer therapeutic heterogeneity in single-cell RNA-seq data. Genome Med. 13, 187 (2021).
Yang, H. et al. Pharmacogenomic profiling of intra-tumor heterogeneity using a large organoid biobank of liver cancer. Cancer Cell 42, 535–551.e8 (2024).
Zhao, W. et al. Deconvolution of cell type-specific drug responses in human tumor tissue with single-cell RNA-seq. Genome Med. 13, 82 (2021).
Hsieh, C.-Y. et al. scDrug: from single-cell RNA-seq to drug response prediction. Comput. Struct. Biotechnol. J. 21, 150–157 (2023).
Ianevski, A. et al. Single-cell transcriptomes identify patient-tailored therapies for selective co-inhibition of cancer clones. Nat. Commun. 15, 8579 (2024).
Tang, C. et al. Personalized tumor combination therapy optimization using the single-cell transcriptome. Genome Med. 15, 105 (2023).
Califano, A. & Alvarez, M. J. The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat. Rev. Cancer 17, 116–130 (2017).
Carro, M. S. et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 463, 318–325 (2010).
Paull, E. O. et al. A modular master regulator landscape controls cancer transcriptional identity. Cell 184, 334–351.e20 (2021).
Aytes, A. et al. Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer Cell 25, 638–651 (2014).
Zeleke, T. Z. et al. Network-based assessment of HDAC6 activity predicts preclinical and clinical responses to the HDAC6 inhibitor ricolinostat in breast cancer. Nat. Cancer 4, 257–275 (2022).
Alvarez, M. J. et al. A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat. Genet. 50, 979–989 (2018).
Mundi, P. S. et al. A transcriptome-based precision oncology platform for patient–therapy alignment in a diverse set of treatment-resistant malignancies. Cancer Discov. 13, 1386–1407 (2023).
Margolin, A. A. et al. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 7, S7 (2006).
Lachmann, A., Giorgi, F. M., Lopez, G. & Califano, A. ARACNe-AP: gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics 32, 2233–2235 (2016).
Filbin, M. G. et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335 (2018).
Liu, I. et al. The landscape of tumor cell states and spatial organization in H3-K27M mutant diffuse midline glioma across age and location. Nat. Genet. 54, 1881–1894 (2022).
Buczkowicz, P. et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 46, 451–456 (2014).
Panditharatna, E., Yaeger, K., Kilburn, L. B., Packer, R. J. & Nazarian, J. Clinicopathology of diffuse intrinsic pontine glioma and its redefined genomic and epigenomic landscape. Cancer Genet. 208, 367–373 (2015).
Lapin, D. H., Tsoli, M. & Ziegler, D. S. Genomic insights into diffuse intrinsic pontine glioma. Front. Oncol. 7, 57 (2017).
Schwartzentruber, J. et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012).
Wu, G. et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 46, 444–450 (2014).
Hoffman, L. M. et al. Clinical, radiologic, pathologic, and molecular characteristics of long-term survivors of diffuse intrinsic pontine glioma (DIPG): a collaborative report from the International and European Society for Pediatric Oncology DIPG Registries. J. Clin. Oncol. 36, 1963–1972 (2018).
Kaplan, A. M. et al. Brainstem gliomas in children. A children’s cancer group review of 119 cases. Pediatr. Neurosurg. 24, 185–192 (1996).
Lin, G. L. et al. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci. Transl. Med. 11, eaaw0064 (2019).
Findlay, I. J. et al. Pharmaco-proteogenomic profiling of pediatric diffuse midline glioma to inform future treatment strategies. Oncogene 41, 461–475 (2022).
Noon, A. & Galban, S. Therapeutic avenues for targeting treatment challenges of diffuse midline gliomas. Neoplasia 40, 100899 (2023).
Mayr, L. et al. Effective targeting of PDGFRA-altered high-grade glioma with avapritinib. Cancer Cell https://doi.org/10.1016/j.ccell.2025.02.018 (2025).
Hoffman, L. M. et al. Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathol. Commun. 4, 1 (2016).
Jones, C. & Baker, S. J. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat. Rev. Cancer 14, 651–661 (2014).
Grasso, C. S. et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 21, 555–559 (2015).
Nagaraja, S. et al. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell 31, 635–652.e6 (2017).
Ding, H. et al. Quantitative assessment of protein activity in orphan tissues and single cells using the metaVIPER algorithm. Nat. Commun. 9, 1471 (2018).
Alvarez, M. J. et al. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 48, 838–847 (2016).
Basso, K. et al. Reverse engineering of regulatory networks in human B cells. Nat. Genet. 37, 382–390 (2005).
Patel, A. P. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014).
Obradovic, A. et al. Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell 184, 2988–3005.e16 (2021).
Magri, L. et al. c-Myc-dependent transcriptional regulation of cell cycle and nucleosomal histones during oligodendrocyte differentiation. Neuroscience 276, 72–86 (2014).
Pozniak, P. D., White, M. K. & Khalili, K. TNF-α/NF-κB signaling in the CNS: possible connection to EPHB2. J. Neuroimmune Pharmacol. 9, 133–141 (2014).
Wang, S., Wang, Y. & Zou, S. A glance at the molecules that regulate oligodendrocyte myelination. Curr. Issues Mol. Biol. 44, 2194–2216 (2022).
Genoud, S. et al. Notch1 control of oligodendrocyte differentiation in the spinal cord. J. Cell Biol. 158, 709–718 (2002).
Jeffries, M. A. et al. mTOR signaling regulates metabolic function in oligodendrocyte precursor cells and promotes efficient brain remyelination in the cuprizone model. J. Neuroscience 41, 8321–8337 (2021).
Lebrun-Julien, F. et al. Balanced mTORC1 activity in oligodendrocytes is required for accurate CNS myelination. J. Neurosci. 34, 8432–8448 (2014).
Čokić, V. P. et al. Proinflammatory cytokine IL-6 and JAK–STAT signaling pathway in myeloproliferative neoplasms. Mediators Inflamm. 2015, 453020 (2015).
WANG, T. et al. The role of the JAK–STAT pathway in neural stem cells, neural progenitor cells and reactive astrocytes after spinal cord injury. Biomed. Rep. 3, 141–146 (2015).
Okada, S. et al. Blockade of interleukin-6 receptor suppresses reactive astrogliosis and ameliorates functional recovery in experimental spinal cord injury. J. Neurosci. Res. 76, 265–276 (2004).
Kummer, K. K., Zeidler, M., Kalpachidou, T. & Kress, M. Role of IL-6 in the regulation of neuronal development, survival and function. Cytokine 144, 155582 (2021).
Katzenellenbogen, B. S., Guillen, V. S. & Katzenellenbogen, J. A. Targeting the oncogenic transcription factor FOXM1 to improve outcomes in all subtypes of breast cancer. Breast Cancer Res. 25, 76 (2023).
Luo, Y. et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 48, D882–D889 (2020).
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Alvarez, M. J. et al. Reply to 'H-STS, L-STS and KRJ-I are not authentic GEPNET cell lines'. Nat. Genet. 51, 1427–1428 (2019).
Bush, E. C. et al. PLATE-seq for genome-wide regulatory network analysis of high-throughput screens. Nat. Commun. 8, 105 (2017).
Vasciaveo, A. et al. OncoLoop: a network-based precision cancer medicine framework. Cancer Discov. 13, 386–409 (2023).
Lugowska, I., Koseła-Paterczyk, H., Kozak, K. & Rutkowski, P. Trametinib: a MEK inhibitor for management of metastatic melanoma. Onco. Targets Ther. 8, 2251–2259 (2015).
Uba, A. İ & Yelekçi, K. Exploration of the binding pocket of histone deacetylases: the design of potent and isoform-selective inhibitors. Turk. J. Biol. 41, 901–918 (2017).
Kumar, S. K. et al. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood 125, 443–448 (2015).
Cattrini, C., Capaia, M., Boccardo, F. & Barboro, P. Etoposide and topoisomerase II inhibition for aggressive prostate cancer: data from a translational study. Cancer Treat. Res. Commun. 25, 100221 (2020).
Schwark, K. et al. Receptor tyrosine kinase (RTK) targeting in pediatric high-grade glioma and diffuse midline glioma: pre-clinical models and precision medicine. Front. Oncol. 12, 922928 (2022).
Waggoner, M. M., Katsetos, P.-C. J., Thomas, M. M. P.-C. E., Galinsky, B. M. A.-C. I. & Fox, P.-C. H. Practical management of the venetoclax-treated patient in chronic lymphocytic leukemia and acute myeloid leukemia. J. Adv. Pract. Oncol. 13, 400–415 (2022).
Federman, N. & McDermott, R. Larotrectinib, a highly selective tropomyosin receptor kinase (TRK) inhibitor for the treatment of TRK fusion cancer. Expert Rev. Clin. Pharmacol. 12, 931–939 (2019).
Han, D. et al. Napabucasin, a novel STAT3 inhibitor suppresses proliferation, invasion and stemness of glioblastoma cells. J. Exp. Clin. Cancer Res. 38, 289 (2019).
Hennika, T. et al. Pre-clinical study of panobinostat in xenograft and genetically engineered murine diffuse intrinsic pontine glioma models. PLoS ONE 12, e0169485 (2017).
D’Amico, R. et al. The addition of sunitinib to radiation delays tumor growth in a murine model of glioblastoma. Neurol. Res. 34, 252–261 (2012).
Ianevski, A., Giri, A. K. & Aittokallio, T. SynergyFinder 3.0: an interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 50, W739–W743 (2022).
Rusert, J. M. et al. Functional precision medicine identifies new therapeutic candidates for medulloblastoma. Cancer Res. 80, 5393–5407 (2020).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Huynh-Thu, V. A., Irrthum, A., Wehenkel, L. & Geurts, P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 5, e12776 (2010).
Chan, T. E., Stumpf, M. P. H. & Babtie, A. C. Gene regulatory network inference from single-cell data using multivariate information measures. Cell Syst. 5, 251–267.e3 (2017).
Skok Gibbs, C. et al. High-performance single-cell gene regulatory network inference at scale: the Inferelator 3.0. Bioinformatics 38, 2519–2528 (2022).
Kamimoto, K. et al. Dissecting cell identity via network inference and in silico gene perturbation. Nature 614, 742–751 (2023).
Teschendorff, A. E. & Wang, N. Improved detection of tumor suppressor events in single-cell RNA-seq data. NPJ Genom. Med. 5, 43 (2020).
Schlauch, D., Paulson, J. N., Young, A., Glass, K. & Quackenbush, J. Estimating gene regulatory networks with pandaR. Bioinformatics 33, 2232–2234 (2017).
Rajbhandari, P. et al. Cross-cohort analysis identifies a TEAD4–MYCN positive feedback loop as the core regulatory element of high-risk neuroblastoma. Cancer Discov. 8, 582–599 (2018).
Jamison, J. K. et al. Entinostat in patients with relapsed or refractory abdominal neuroendocrine tumors. Oncologist 29, 817–e1213 (2024).
Ziegler, D. S. et al. Brief report: potent clinical and radiological response to larotrectinib in TRK fusion-driven high-grade glioma. Br. J. Cancer 119, 693–696 (2018).
Laetsch, T. W. et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 19, 705–714 (2018).
Obradovic, A. et al. Systematic elucidation and pharmacological targeting of tumor-infiltrating regulatory T cell master regulators. Cancer Cell 41, 933–949.e11 (2023).
Palmer, A. C. & Sorger, P. K. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell 171, 1678–1691.e13 (2017).
Riess, C. et al. Cyclin-dependent kinase inhibitors in head and neck cancer and glioblastoma—Backbone or add-on in immune-oncology? Cancer Metastasis Rev. 40, 153–171 (2021).
Liu, G. H., Chen, T., Zhang, X., Ma, X. L. & Shi, H. S. Small molecule inhibitors targeting the cancers. MedComm 3, e181 (2022).
Gerrits, E., Heng, Y., Boddeke, E. W. G. M. & Eggen, B. J. L. Transcriptional profiling of microglia; current state of the art and future perspectives. Glia 68, 740–755 (2020).
Pacific Pediatric Neuro-Oncology Consortium (PNOC). https://pnoc.us
The Childhood Brain Tumor Tissue Consortium (CBTTC). https://cbtn.org
Vlahos, L. et al. Systematic, protein activity-based characterization of single cell state. Preprint at https://doi.org/10.1101/2021.05.20.445002 (2023).
Douglass, E. F. et al. A community challenge for a pancancer drug mechanism of action inference from perturbational profile data. Cell Rep. Med. 3, 100492 (2022).
Woo, J. H. et al. Elucidating compound mechanism of action by network perturbation analysis. Cell 162, 441–451 (2015).
Schlattmann, P. Medical Applications of Finite Mixture Models (Springer, 2009).
Do, C. B. & Batzoglou, S. What is the expectation maximization algorithm? Nat. Biotechnol. 26, 897–899 (2008).
Acknowledgements
We thank M. Monje for generously sharing the SU-DIPG-IV (DIPG4), SU-DIPG-VI (DIPG6), SU-DIPG-XVII (DIPG17) and SU-DIPG-XXXVI (DIPG36) cell lines. We thank R. Hashizume for sharing the SF8628 and SF7761 cell lines. We thank O. Becher for sharing the DIPG4423 cell line. Lastly, we thank the Columbia Genome Center, High-Throughput Screening and Oncology Precision Therapeutics and Imaging Core (OPTIC) cores for their help. This research was primarily supported by generous funding from Swim Across America (J.P., R.D.G. and C-C.W.), The Team Jack Foundation (J.P.), Hyundai Hope on Wheels Impact Award (S.Z. and J.P.) and the Gary and Yael Fegel Family Foundation (S.Z., J.P., L.S. and C-C.W.). Additional funding support was provided by the Columbia University Irving Medical Center Herbert and Florence Irving Basic Science Scholar award and Pediatric Oncology Novel Research Fund Award awarded to J.P., as well as the Vagelos College of Physicians and Surgeons Interdisciplinary Research Initiatives Seed (IRIS) Fund (J.P., C-C.W. and A.C.). E.C.F. was also supported by the Postgraduate studies in North America and the Asia-Pacific Region Fellowship by ‘La Caixa Foundation’ and the Trainee Associate Member (TAM) predoctoral award from the Herbert Irving Comprehensive Cancer and Education Coordination centers at Columbia University. T.T. was supported by fellowships from the German Research Foundation and ChadTough Defeat DIPG Foundation. R.D.G. was also supported by the Hyundai Hope on Wheels Hope Scholar Award, Rally Foundation, StacheStrong and the Musella Foundation. C-C.W. was also supported by the Sebastian Strong Foundation, the St. Baldrick’s Foundation Scholar Award and the Pediatric Cancer Foundation. M.G. was partially supported by an ASCO Young Investigator Award as well as a grant from the Radiological Society of North America (RSNA). A.T.G. was supported, in part, by the Ruth L. Kirschstein National Research Service Award (NRSA) Institutional Research Training Grant (5T32GM007367-43), and in part by the National Cancer Institute (NCI) Ruth L. Kirschstein National Research Service Award Individual Fellowship (F30CA257765). Additional support for this work was provided by the NCI Outstanding Investigator award R35 CA 197745, the National Institutes of Health (NIH) Shared Instrumentation Grants S10 OD012351 and S10 OD021764, and U01CA272610 (all to A.C.), the National Institute of Neurological Disorders and Stroke (NINDS) Research Program Award R35 NS122339 (to R.W.-R.), NIH Grants R01 CA277605-01A1 and R01 NS132344-01 to Z.Z, NIH Grant P30 CA013696 (NCI) supporting use of OPTIC and the NIH/NCI Cancer Center Support Grant P30CA013696 supporting use of the Genomics and High-Throughput Screening Shared Resource, and by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1TR001873. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
J.P., A.C. and E.C.F. designed the study and wrote the manuscript. S.Z. supported study conception and collaborative coordination. J.P. led the execution and integration of computational analyses across the study. E.C.F. conducted and analyzed experimental and animal studies and contributed broadly to all computational analyses, including leading CRISPR–Cas9 screen, subcutaneous murine model and cell line single-cell dataset analyses. L.T. contributed to computational analyses, including for syngeneic orthotopic DMG model single-cell data, and managed the associated data repository. J.W. and A.O. contributed to the computational analysis of the DMG patient single-cell data, including single-cell OncoTarget and OncoTreat analyses. X.Z. conducted CRISPR–Cas9 screening experiments under the supervision of Z.Z. L.V. contributed to procurement and analysis of single-cell data from patients with DMG. P.L. and A.T.G. contributed to the computational analysis of the CRISPR–Cas9 screen and ARACNe network inference, respectively. H.M., D.V.M., C.S., M.G. and H-J.W. supported animal experiments and imaging. T.J.M., P.S.B., J.R.C., T.T. and R.W-R. generated patient bulk RNA-seq data and performed drug screening assays. R.D.G. and C-C.W. provided resources and guidance for animal model development and experiments. L.S., J.G. and S.Z. contributed clinical expertise and guidance on patient cohorts and therapeutic prioritization. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
A.C. is a founder, equity holder and consultant of DarwinHealth, a company that has licensed some of the algorithms used in this paper from Columbia University. Columbia University is also an equity holder in DarwinHealth. P.L. and L.T. work at DarwinHealth. S.Z. was the senior director of pediatric oncology at Bristol Myers Squibb during this study, but no longer holds that position. P.S.B. is a consultant at Accordant Health Services and has received research funding from GPCR Therapeutics. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Aaron Diaz, Sven Nelander and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Validation of diffuse midline glioma cell states and master regulators in an independent single-cell RNA sequencing dataset.
a, UMAP projection of VIPER-inferred protein activity across 1,629 tumor cells from an independent cohort of eight pediatric diffuse midline glioma (DMG) patient samples, with cells colored by clusters identified using resolution-optimized Louvain clustering. b, UMAP from (a) with cells annotated in a supervised manner based on statistically significant enrichment of previously defined human DMG cell state master regulators (MRs) within the VIPER-inferred protein activity signature of each cell (one-sided Gene Set Enrichment Analysis (GSEA), FDR ≤ 0.05), demonstrating strong concordance between unsupervised clustering and supervised cell state assignment. c, Distribution of the proportion of cells assigned to each DMG cell state (y-axis) across the eight patient tumors (x-axis), showing that all cell states are represented within each tumor. d, Heatmap of viperSimilarity normalized enrichment scores (NES) quantifying concordance between cell state MRs identified in the initial six-patient discovery dataset (D1; 3,032 tumor cells) and the independent eight-patient validation dataset (D2; 1,629 tumor cells). Red indicates concordant enrichment; blue indicates inverse enrichment. e, UMAP projections of VIPER-inferred protein activity from an integrated analysis of 4,661 tumor cells across all 14 DMG patients from datasets D1 and D2. Resolution-optimized Louvain clustering identified eight clusters (top), which were subsequently assigned to the seven previously defined DMG cell states based on lineage-marker protein activity (bottom). f, Heatmap of Stouffer-integrated protein activity (NES) for oligodendrocyte precursor cell (OPC), oligodendrocyte (OC), and astrocyte (AC) lineage markers, as well as proliferative markers (rows), across the eight unsupervised clusters from the integrated analysis (columns), supporting cluster annotation. g, Heatmap of viperSimilarity enrichment scores (NES) quantifying concordance between cell state MRs across all 14 individual patient samples, shown as a color gradient from blue (inverse enrichment) to red (concordant enrichment), demonstrating highly conserved regulatory programs across tumors. Cell state annotations and patient identity are indicated by the horizontal and vertical bars. h, Heatmap of viperSimilarity enrichment scores (NES) quantifying concordance between cell state MRs inferred from DMG single-cell regulatory networks and those derived from the DMG bulk regulatory network. Red indicates concordant enrichment and blue indicates inverse enrichment.
Extended Data Fig. 2 CRISPR validation of VIPER-inferred tumorigenic master regulators in diffuse midline glioma cell line models profiled by single-cell RNA sequencing.
a, One-sided Gene Set Enrichment Analysis (GSEA) showing enrichment of CRISPR-essential genes (defined by log2FC ≤ 0 and FDR ≤ 0.05; red ticks) within the VIPER-inferred synthetic bulk tumorigenic protein activity signature of human diffuse midline glioma (DMG) malignant cells (GTEx caudate reference) across three DMG cell lines (DIPG17, DIPG4, SF8628). Normalized enrichment scores (NES) and p-values are shown for each line: DIPG17 (p = 5×10−3), DIPG4 (p = .027), and SF8628 (p = 0.01). b, FDR-adjusted p-values from one-sided GSEA assessing enrichment of CRISPR-essential genes identified in each DMG line and in the integrated set across all three lines, evaluated against synthetic bulk tumorigenic protein activity signatures for each of the seven DMG cell states. Oligodendrocyte precursor cell (OPC)-like tumorigenic master regulators (MRs) showed the most consistent enrichment, while SF8628-specific essential genes uniquely showed enrichment for astrocyte (AC)-like tumorigenic MRs. c, FDR-adjusted p-values from one-sided GSEA testing enrichment of CRISPR-essential genes within synthetic bulk tumorigenic protein activity signatures derived from malignant cells in each of the six patient samples profiled by single-cell RNA sequencing (scRNA-seq). DIPG4-specific essential genes uniquely enriched in the MUV5 patient sample, which shares the HIST1H3B mutation. d, Heatmap of patient-specific synthetic bulk tumorigenic protein activity for 37 CRISPR-validated essential MRs (rows) comprising the leading edge of enrichment in at least one DMG patient sample (columns). The annotation bar indicates patient-specific enrichment based on one-sided GSEA (black, FDR ≤ 0.05; gray, not significant). e, One-sided GSEA p-values for enrichment of essential genes from each CRISPR-screened cell line within tumorigenic protein activity signatures of six DMG cell lines profiled by bulk RNA-seq, supporting VIPER’s ability to nominate sample-specific essential MRs. f, UMAP projection of VIPER-inferred protein activity across 11,325 single cells from six DMG patient-derived cell lines profiled by scRNA-seq. Cell numbers and genotypes (histone and TP53 mutation status) are summarized below. g, UMAP from (f) colored by assigned human DMG cell state based on strongest statistically significant enrichment of state-specific MRs per cell (one-sided GSEA, FDR-adjusted). Cells not meeting significance (FDR > 0.05) are labeled unassigned, reflecting cell line-specific dependencies driven by ex vivo culture conditions.
Extended Data Fig. 3 Pharmacodynamic validation of OPC-like master regulator inversion by OncoTarget/OncoTreat-predicted drugs in a DIPG17 cell line-derived subcutaneous DMG xenograft.
Analytic Rank-based Enrichment Analysis (aREA) plots showing enrichment of the top (red ticks) and bottom (blue ticks) 50 human diffuse midline glioma (DMG) cell state-specific tumorigenic MRs within differential protein activity signatures comparing drug- versus vehicle-treated single cells from the same state obtained after five days of treatment. For each drug, enrichment is shown separately for oligodendrocyte precursor cell (OPC)-like and astrocyte (AC)-like cells. All five OPC-targeting drugs (avapritinib, dinaciclib, trametinib, mocetinostat, and etoposide) significantly inverted patient-derived OPC tumorigenic MRs in OPC-like cells, reflected by negative aREA normalized enrichment scores (NES), with no corresponding inversion in AC-like cells. P-values correspond to two-sided aREA for each drug-cell state comparison.
Extended Data Fig. 4 Pharmacodynamic validation of AC-like master regulator inversion by OncoTarget/OncoTreat-predicted drugs in a DIPG17 cell line-derived subcutaneous DMG xenograft.
a, Analytic Rank-based Enrichment Analysis (aREA) plots show enrichment of the top (red ticks) and bottom (blue ticks) 50 human diffuse midline glioma (DMG) cell state-specific tumorigenic MRs within differential protein activity signatures comparing drug- versus vehicle-treated single cells from the same state obtained after five days of treatment. For each drug, enrichment is shown separately for astrocyte (AC)-like states (AC-like and OPC/AC-like) and oligodendrocyte precursor cell (OPC)-like states. Three of the four astrocyte (AC)-targeting drugs (ruxolitinib, venetoclax, and larotrectinib) significantly inverted patient-derived AC tumorigenic MRs in AC-like cells, reflected by negative aREA normalized enrichment score (NES), with weak to no corresponding inversion in OPC-like cells, except for napabucasin which significantly inverted only OPC-like tumorigenic MRs in OPC-like cells in line with its OPC state-selective depletion. P-values correspond to two-sided aREA for each drug-cell state comparison. b, Cell state-specific VIPER-inferred differential protein activity analyses confirmed drug-mediated inversion of NTRK2 activity (NES, x-axis) by larotrectinib in oligodendrocyte (OC)-like and OPC/AC-like cell states (cell states evaluated along y-axis). c, Differential VIPER-inferred protein activity analysis of STAT3 activity (NES, x-axis) following napabucasin treatment in AC_Integrated (AC and OPC/AC) and OPC_Integrated (OPCQ, OPC, OPCC) cells (y-axis) showed no inversion of STAT3 activity, indicating lack of effective pharmacodynamic target engagement across states by napabucasin in this model. Abbreviations: OPCQ, OPC Quiescent; OPCC, OPC Cycling.
Extended Data Fig. 5 Generation and single-cell validation of a DIPG4423 syngeneic orthotopic diffuse midline glioma murine model.
a-b, Representative magnetic resonance imaging (MRI) scans of the brainstem in DIPG4423-injected mice showing a, focal enhancement at the injection site two weeks post-injection (prior to treatment; pink arrow), consistent with tumor establishment, and b, tumor progression three weeks following vehicle treatment. c, UMAP projection of VIPER-inferred protein activity from single-cell RNA sequencing (scRNA-seq) of two independent DIPG4423 orthotopic tumors (red and green) and normal murine brainstem tissue (blue). Immune cells, annotated using singleR, were excluded from all analyses. d, InferCNV analysis comparing non-immune cells from the two tumor samples to the untreated wild-type mouse brainstem identified clusters with extensive copy number alterations, delineating malignant cell populations (n = 8,013 single cells). e, UMAP projection of VIPER-inferred protein activity across malignant cells demonstrated minimal batch effect between independently generated tumors. f, Same UMAP as in (e), with cells colored by unsupervised clusters identified by resolution-optimized Louvain clustering. Unsupervised clusters corresponded closely to supervised human diffuse midline glioma (DMG) cell state assignments based on enrichment of human state-specific master regulators (MRs; see Fig. 5d) per cell, supporting preservation of tumor cell state heterogeneity in the orthotopic model. g, Heatmap of viperSimilarity normalized enrichment scores (NES) quantifying concordance between human DMG and orthotopic mouse cell state MRs. Red indicates concordant enrichment and blue indicates inverse enrichment.
Extended Data Fig. 6 Longitudinal MRI and survival analyses demonstrate significant benefit from avapritinib, trametinib, and dinaciclib monotherapy in a DIPG4423 syngeneic orthotopic diffuse midline glioma model.
a, Longitudinal magnetic resonance imaging (MRI) of representative DIPG4423 syngeneic orthotopic diffuse midline glioma (DMG) tumor-bearing mice treated with monotherapies that conferred a significant survival advantage relative to vehicle control (avapritinib, trametinib, dinaciclib). Each column represents a treatment arm, and each row corresponds to an imaging time point from the same mouse. “X” indicates animals that reached endpoint due to tumor progression prior to the indicated time point. b, Kaplan-Meier survival analyses comparing each monotherapy arm individually with vehicle-treated controls, demonstrating significantly prolonged survival with (top to bottom) avapritinib (n = 4, p = 1.9×10−3), trametinib (n = 5, p = 0.03), and dinaciclib (n = 5, p = 3.3×10−3), relative to vehicle (n = 9). Statistical significance was assessed using the log-rank test. Asterisks denote significance: *p < 0.05, **p < 0.01.
Extended Data Fig. 7 Longitudinal MRI of trametinib-based combinations and tumor volume reduction across all combination therapies in a DIPG4423 syngeneic orthotopic diffuse midline glioma model.
a, Longitudinal magnetic resonance imaging (MRI) of representative DIPG4423 syngeneic orthotopic diffuse midline glioma (DMG) tumor-bearing mice treated with trametinib monotherapy or trametinib + ruxolitinib combination therapy, demonstrating delayed tumor progression relative to vehicle control and enhanced tumor control with combination treatment compared to trametinib alone. Each column represents a treatment condition, with images shown from two individual mice (biological replicates) per treatment arm at the indicated time points (rows). “X” denotes animals that reached endpoint due to tumor progression prior to the indicated imaging time point. b, Tumor volumes (y-axis) measured two weeks after treatment initiation across all combination therapy arms compared with vehicle control (y-axis). Each dot represents an individual mouse tumor (biological replicate); bars indicate mean ± SEM (standard error of the mean). Sample sizes and statistical significance (two-sided Welch’s t-test, relative to vehicle) were as follows: trametinib + venetoclax (n = 5, p = 3.7×10−3), avapritinib + venetoclax (n = 5, p < 1×10−4), mocetinostat + venetoclax (n = 5, p = 1.9×10−3), avapritinib + ruxolitinib (n = 3, p < 1×10−4), dinaciclib + ruxolitinib (n = 3, p = 2×10−4), trametinib + ruxolitinib (n = 4, p < 1×10−4), and avapritinib + larotrectinib (n = 3, p = 1×10−4). Vehicle control n = 9.
Extended Data Fig. 8 Longitudinal MRI of dinaciclib-based combinations showing enhanced tumor control in a DIPG4423 syngeneic orthotopic diffuse midline glioma murine model.
Longitudinal magnetic resonance imaging (MRI) of representative DIPG4423 syngeneic orthotopic diffuse midline glioma (DMG) tumor-bearing mice treated with dinaciclib monotherapy or dinaciclib + ruxolitinib combination therapy, compared with vehicle control. Dinaciclib + ruxolitinib treatment showed delayed tumor progression and prolonged radiographic tumor control relative to both vehicle-treated mice and dinaciclib monotherapy. Each column represents a treatment condition, with images shown from two individual mice (biological replicates) per treatment arm at the indicated time points (rows). “X” denotes animals that reached endpoint due to tumor progression prior to the indicated imaging time point.
Extended Data Fig. 9 Longitudinal MRI of avapritinib-based combinations showing enhanced tumor control in a DIPG4423 syngeneic orthotopic diffuse midline glioma murine model.
Longitudinal magnetic resonance imaging (MRI) of representative DIPG4423 syngeneic orthotopic diffuse midline glioma (DMG) tumor-bearing mice treated with avapritinib monotherapy or avapritinib-based combination therapies, including avaptinib + venetoclax, avapritinib + larotrectinib, and avapritinib + ruxolitinib, compared with vehicle control. Avapritinib-based combinations showed delayed tumor progression and prolonged radiographic tumor control relative to both vehicle-treated mice and avapritinib monotherapy. Each column represents a treatment condition, with images shown from two individual mice (biological replicates) per treatment arm at the indicated time points (rows). “X” denotes animals that reached endpoint due to tumor progression prior to the indicated imaging time point.
Extended Data Fig. 10 BLISS independence analysis indicating that in vivo synergy of avapritinib/ruxolitinib combination therapy is not driven by cell-autonomous drug synergy in the in vitro predominant OPC-like cell state.
a, BLISS independence analysis of avapritinib + ruxolitinib combination treatment in three diffuse midline glioma (DMG) cell lines—DIPG36 (human, HIST1H3B), DIPG4423 (murine, H3F3A), and SF8628 (human, H3F3A)—showed predominantly additive effects, with Bliss synergy scores (δ) largely within the −10 to +10 range, indicating absence of measurable drug synergy in vitro. b, Dose-response curves for ruxolitinib monotherapy across the three DMG cell lines showing limited in vitro efficacy, with DIPG36 cells (left) exhibiting continued proliferation across the tested concentration range on treatment. c, Dose-response curves for avapritinib monotherapy across the same cell lines showing consistent in vitro sensitivity, with IC50 < 2μM in all three models.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–7 (figures and figure legends).
Supplementary Tables (download XLSX )
Supplementary Table 1. DMG cell state differentially expressed genes and top 50 cell state master regulators. Supplementary Table 2. Clinical and molecular characteristics of DMG bulk RNA-seq cohorts from the St. Jude, PNOC and CBTTC datasets used to generate the bulk regulatory network. Supplementary Table 3. Pooled CRISPR–Cas9 knockout screen transcription factor sgRNA library, internal controls, read counts and MAGeCK analysis outputs. Supplementary Table 4. PLATE-seq drug perturbation metadata (SF8628 and DIPG6) and OncoTreat and OncoTarget DMG cell state-specific normalized enrichment scores (NES). Supplementary Table 5. Drug-induced changes in tumor cell state composition in DIPG17 subcutaneous DMG models by scRNA-seq and 2 week tumor volumes in DIPG4423 orthotopic DMG models.
Source data
Source Data Fig. 5 (download XLSX )
Source Data for Fig. 5h,k. Two-week tumor volumes in DIPG4423 orthotopic DMG models, both for monotherapies and all combination therapies.
Source Data Extended Fig. 7 (download XLSX )
Source Data for Extended Data Fig. 7b. Two-week tumor volumes in DIPG4423 orthotopic DMG model,s both for monotherapies and all combination therapies.
Source Data Fig. 4 (download XLSX )
Source Data for Fig. 4f,g. Number of cells assigned to each cell state by drug-treated biological replicate profiled by scRNA-seq (DIPG17 subcutaneous xenograft tumors).
Source Data Supplementary Fig. 4 (download XLSX )
Source Data for Supplementary Fig. 4. Number of cells assigned to each cell state by drug-treated biological replicate profiled by scRNA-seq (DIPG17 subcutaneous xenograft tumors).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Calvo Fernández, E., Tomassoni, L., Zhang, X. et al. Systematic design of combination therapy by targeting master regulators of coexisting diffuse midline glioma cell states. Nat Genet (2026). https://doi.org/10.1038/s41588-026-02550-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41588-026-02550-w
