
Abstract
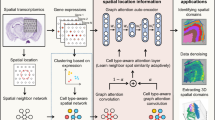
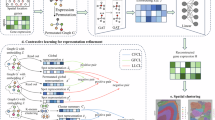
Spatially resolved transcriptomics (SRT) technologies have significantly advanced biomedical research, but their data analysis remains challenging due to the discrete nature of the data and the high levels of noise, compounded by complex spatial dependencies. Here, we propose spaVAE, a dependency-aware, deep generative spatial variational autoencoder model that probabilistically characterizes count data while capturing spatial correlations. spaVAE introduces a hybrid embedding combining a Gaussian process prior with a Gaussian prior to explicitly capture spatial correlations among spots. It then optimizes the parameters of deep neural networks to approximate the distributions underlying the SRT data. With the approximated distributions, spaVAE can contribute to several analytical tasks that are essential for SRT data analysis, including dimensionality reduction, visualization, clustering, batch integration, denoising, differential expression, spatial interpolation, resolution enhancement and identification of spatially variable genes. Moreover, we have extended spaVAE to spaPeakVAE and spaMultiVAE to characterize spatial ATAC-seq (assay for transposase-accessible chromatin using sequencing) data and spatial multi-omics data, respectively.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

Data availability
All data supporting the findings of the study are deposited and available at https://doi.org/10.6084/m9.figshare.21623148.v5 (ref. 68).
Code availability
An open-source software implementation of spaVAE, spaPeakVAE, spaMultiVAE, spaLDVAE and spaPeakLDVAE is available on GitHub: https://github.com/ttgump/spaVAE. It is also available in the Zenodo repository69.
References
Asp, M., Bergenstrahle, J. & Lundeberg, J. Spatially resolved transcriptomes: next generation tools for tissue exploration. Bioessays 42, e1900221 (2020).
Rao, A., Barkley, D., Franca, G. S. & Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 596, 211–220 (2021).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Eraslan, G., Simon, L. M., Mircea, M., Mueller, N. S. & Theis, F. J. Single-cell RNA-seq denoising using a deep count autoencoder. Nat. Commun. 10, 390 (2019).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Tian, T., Wan, J., Song, Q. & Wei, Z. Clustering single-cell RNA-seq data with a model-based deep learning approach. Nat. Mach. Intell. 1, 191–198 (2019).
Tian, T., Zhang, J., Lin, X., Wei, Z. & Hakonarson, H. Model-based deep embedding for constrained clustering analysis of single cell RNA-seq data. Nat. Commun. 12, 1873 (2021).
Hu, J. et al. SpaGCN: integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 18, 1342–1351 (2021).
Long, Y. et al. Spatially informed clustering, integration, and deconvolution of spatial transcriptomics with GraphST. Nat. Commun. 14, 1155 (2023).
Dong, K. & Zhang, S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat. Commun. 13, 1739 (2022).
Lin, X., Gao, L., Whitener, N., Ahmed, A. & Wei, Z. A model-based constrained deep learning clustering approach for spatially resolved single-cell data. Genome Res. 32, 1906–1917 (2022).
Pham, D. et al. stLearn: integrating spatial location, tissue morphology and gene expression to find cell types, cell-cell interactions and spatial trajectories within undissociated tissues. Nat Commun. 14, 7739 (2023).
Shang, L. & Zhou, X. Spatially aware dimension reduction for spatial transcriptomics. Nat. Commun. 13, 7203 (2022).
Zhao, E. et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 39, 1375–1384 (2021).
Casale, F. P., Dalca, A. V., Saglietti, L., Listgarten, J. & Fusi, N. Gaussian process prior variational autoencoders. In Proc. 32nd International Conference on Neural Information Processing Systems (NIPS 2018) (eds Bengio, S. et al.) (Curran Associates, Inc., 2018).
Kingma, D. P. & Welling, M. Auto-encoding variational Bayes. In International Conference on Learning Representations (2013).
Titsias, M. Variational learning of inducing variables in sparse Gaussian processes. Proceedings of Machine Learning Research 5, 567–574 (2009).
Hensman, J., Fusi, N. & Lawrence, N. D. Gaussian processes for big data. In Proc. 29th Conference on Uncertainty in Artificial Intelligence (UAI 2013) (eds Nicholson, A. & Smyth, P.) (AUAI Press, 2013).
Jazbec, M. et al. Scalable Gaussian process variational autoencoders. Proceedings of Machine Learning Research 130, 3511–3519 (2021).
Deng, Y. et al. Spatial profiling of chromatin accessibility in mouse and human tissues. Nature 609, 375–383 (2022).
Jiang, F. et al. Simultaneous profiling of spatial gene expression and chromatin accessibility during mouse brain development. Nat. Methods 20, 1048–1057 (2023).
Liu, Y. et al. High-plex protein and whole transcriptome co-mapping at cellular resolution with spatial CITE-seq. Nat. Biotechnol. 41, 1405–1409 (2023).
Liu, Y. et al. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell 183, 1665–1681 (2020).
Xiong, L. et al. SCALE method for single-cell ATAC-seq analysis via latent feature extraction. Nat. Commun. 10, 4576 (2019).
Ashuach, T., Reidenbach, D. A., Gayoso, A. & Yosef, N. PeakVI: a deep generative model for single-cell chromatin accessibility analysis. Cell Rep. Methods 2, 100182 (2022).
Gayoso, A. et al. Joint probabilistic modeling of single-cell multi-omic data with totalVI. Nat. Methods 18, 272–282 (2021).
Maynard, K. R. et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat. Neurosci. 24, 425–436 (2021).
Dries, R. et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 22, 78 (2021).
McInnes, L., Healy, J., Saul, N. & Großberger, L. UMAP: uniform manifold approximation and projection. Journal of Open Source Software 3, 861 (2018).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Stickels, R. R. et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 39, 313–319 (2021).
Lein, E. S. et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 (2007).
Cable, D. M. et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 40, 517–526 (2022).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Svensson, V., Teichmann, S. A. & Stegle, O. SpatialDE: identification of spatially variable genes. Nat. Methods 15, 343–346 (2018).
Sun, S., Zhu, J. & Zhou, X. Statistical analysis of spatial expression patterns for spatially resolved transcriptomic studies. Nat. Methods 17, 193–200 (2020).
Stahl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016).
Andersson, A. et al. Spatial deconvolution of HER2-positive breast cancer delineates tumor-associated cell type interactions. Nat. Commun. 12, 6012 (2021).
Bergenstrahle, L. et al. Super-resolved spatial transcriptomics by deep data fusion. Nat. Biotechnol. 40, 476–479 (2022).
Bravo Gonzalez-Blas, C. et al. cisTopic: cis-regulatory topic modeling on single-cell ATAC-seq data. Nat. Methods 16, 397–400 (2019).
Schep, A. N., Wu, B., Buenrostro, J. D. & Greenleaf, W. J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017).
Dumais, S. T. Latent semantic analysis. Annu. Rev. Inf. Sci. Technol. 38, 188–230 (2005).
Machanick, P. & Bailey, T. L. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697 (2011).
Wong, Y. W. et al. Gene expression analysis of nuclear factor I-A deficient mice indicates delayed brain maturation. Genome Biol. 8, R72 (2007).
Tutukova, S., Tarabykin, V. & Hernandez-Miranda, L. R. The role of neurod genes in brain development, function, and disease. Front. Mol. Neurosci. 14, 662774 (2021).
Higgins, I. et al. beta-VAE: learning basic visual concepts with a constrained variational framework. In International Conference on Learning Representations (2017).
Pearce, M. The Gaussian process prior VAE for interpretable latent dynamics from pixels. Proceedings of Machine Learning Research 118, 1–12 (2020).
Sohn, K., Lee, H. & Yan, X. Learning structured output representation using deep conditional generative models. In Proc. 28th International Conference on Neural Information Processing Systems (NIPS 2015) (eds Cortes, C. et al.) 3483–3491 (MIT Press, 2015).
Ding, J. & Regev, A. Deep generative model embedding of single-cell RNA-seq profiles on hyperspheres and hyperbolic spaces. Nat. Commun. 12, 2554 (2021).
Svensson, V., Gayoso, A., Yosef, N. & Pachter, L. Interpretable factor models of single-cell RNA-seq via variational autoencoders. Bioinformatics 36, 3418–3421 (2020).
Townes, F. W. & Engelhardt, B. E. Nonnegative spatial factorization applied to spatial genomics. Nat. Methods 20, 229–238 (2023).
Paszke, A. et al. Automatic differentiation in PyTorch. In Proc. 31st International Conference on Neural Information Processing Systems (NIPS 2017) (eds Wallach, H. M. et al.) (Curran Associates, Inc., 2017).
Clevert, D.-A., Unterthiner, T. & Hochreiter, S. Fast and accurate deep network learning by exponential linear units (ELUs). In International Conference on Learning Representations (2015).
Ioffe, S. & Szegedy, C. Batch normalization: accelerating deep network training by reducing internal covariate shift. In Proc. 32nd International Conference on International Conference on Machine Learning (ICML 2015) (eds Bach, F. & Blei, D.), Vol. 37, 448–456 (JMLR.org, 2015).
Kingma, D. P. & Ba, J. Adam: a method for stochastic optimization. In 3rd International Conference for Learning Representations (2015).
Loshchilov, I. & Hutter, F. Decoupled weight decay regularization. In International Conference on Learning Representations (2017).
Shao, H. et al. Rethinking controllable variational autoencoders. In 2022 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR) 19228–19237 (IEEE, 2022).
Blondel, V. D., Guillaume, J.-L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008, P10008 (2008).
Boyeau, P. et al. An empirical Bayes method for differential expression analysis of single cells with deep generative models. Proc. Natl Acad. Sci. USA 120, e2209124120 (2023).
Kass, R. E. & Raftery, A. E. Bayes factors. J. Am. Stat. Assoc. 90, 773–795 (1995).
Zhu, J., Sun, S. & Zhou, X. SPARK-X: non-parametric modeling enables scalable and robust detection of spatial expression patterns for large spatial transcriptomic studies. Genome Biol. 22, 184 (2021).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Stuart, T., Srivastava, A., Madad, S., Lareau, C. A. & Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021).
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Tian, T. Spatial genomics datasets. figshare https://doi.org/10.6084/m9.figshare.21623148.v5 (2023).
Tian, T. spaVAE: spatial dependency-aware deep generative models. Zenodo https://doi.org/10.5281/zenodo.8407637 (2023).
Acknowledgements
The study was supported by grant R15HG012087 (Z.W.) from the National Institutes of Health (NIH), grant BK20230781 (J.Z.) from the Natural Science Foundation of Jiangsu Province, and also funded in part by an Institutional Development Fund from The Children’s Hospital of Philadelphia (CHOP) and by CHOP’s Endowed Chair in Genomic Research. This work used the Extreme Science and Engineering Discovery Environment (XSEDE) through the allocation CIE170034, supported by the National Science Foundation grant number ACI1548562. The authors thank R. Cheng from the Tianjin University of Finance and Economics for assistance with the manuscript.
Author information
Authors and Affiliations
Contributions
T.T. conceived the project. T.T. and J.Z. designed the method. T.T., J.Z. and X.L. designed and conducted the experiments. Z.W. and H.H. supervised the study. T.T., J.Z., X.L., Z.W. and H.H. wrote the manuscript. All authors approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks Ofir Lindenbaum and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editors: Hui Hua and Lin Tang, in collaboration with the Nature Methods team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 spaVAE for integrating batches of 10X mouse anterior and posterior brains data.
a, The spaVAE embedding of the mouse brain data with two regions (anterior and posterior) and two batches (section 1 and section 2), with colors and shapes denoting brain regions and batches. b, Clustering labels of the combined four samples, with colors denoting cluster labels. c, Alluvial plot of cluster proportions across different samples. d-e, Top genes identified (log fold change > 1 and Bayes factor > 10) by spaVAE within different clusters among the two sections of mouse anterior brain (d) and among the two sections of mouse posterior brain (e). Heatmaps display relative denoised averaged expression levels across the clusters.
Extended Data Fig. 2 Top spatially and nonspatially variable genes identified by spaLDVAE in the first 6 human DLPFC samples.
Top 4000 highly variable genes are used for this analysis.
Extended Data Fig. 3 Top spatially and nonspatially variable genes identified by spaLDVAE in the last 6 human DLPFC samples.
Top 4000 highly variable genes are used for this analysis.
Supplementary information
Supplementary Information
Supplementary Figs. 1–64, Tables 1 and 2, and Notes 1–14.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tian, T., Zhang, J., Lin, X. et al. Dependency-aware deep generative models for multitasking analysis of spatial omics data. Nat Methods 21, 1501–1513 (2024). https://doi.org/10.1038/s41592-024-02257-y
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41592-024-02257-y
This article is cited by
-
Denoising spatial epigenomic data via deep matrix factorization
Nature Computational Science (2026)
-
spaMGCN: a graph convolutional network with autoencoder for spatial domain identification using multi-scale adaptation
Genome Biology (2025)
-
SMOPCA: spatially aware dimension reduction integrating multi-omics improves the efficiency of spatial domain detection
Genome Biology (2025)
-
DeepGFT: identifying spatial domains in spatial transcriptomics of complex and 3D tissue using deep learning and graph Fourier transform
Genome Biology (2025)
-
SEPAR enables spatial metagene discovery and associated molecular pattern characterization in spatial transcriptomics and multi-omics datasets
Communications Biology (2025)
