
Abstract
Single-cell genomic studies were recently conducted on hundred of samples exhibiting complex designs. These data have tremendous potential for discovering how sample- or tissue-level phenotypes relate to cellular and molecular composition. However, current analyses are often based on simplified representations of these data by averaging information across cells. We present multi-resolution variational inference (MrVI), a deep generative model designed to realize the potential of cohort studies at the single-cell level. MrVI tackles two fundamental, intertwined problems: stratifying samples into groups and evaluating the cellular and molecular differences between groups, without requiring predefined cell states. Leveraging its single-cell perspective, MrVI detects clinically relevant stratifications of cohorts of people with COVID-19 or inflammatory bowel disease that are manifested in only certain cellular subsets, enabling new discoveries that would otherwise be overlooked. MrVI can de novo identify groups of small molecules with similar biochemical properties and evaluate their effects on cellular composition and gene expression in large-scale perturbation studies. MrVI is an open-source tool at scvi-tools.org.
Similar content being viewed by others


Main
Over the past two decades, the use of functional genomics in large-scale, many-sample studies has been instrumental in advancing our understanding of how clinical, genetic and environmental properties manifest at the cellular and molecular levels1,2. These studies now benefit from a potentially transformative increase in quality and resolution, thanks to the maturation of large-scale single-cell genomics, which provides access to detailed information about the cellular and molecular composition of hundreds of samples3,4,5,6,7,8,9. Realizing the potential of large-scale single-cell genomics, however, requires rethinking the analysis strategy. Early on, most studies relied on small numbers of samples and focused on variation between cells; however, the emergence of large-scale single-cell genomics now opens the way for a more in-depth understanding of variation between samples.
There are at least two fundamental tasks in sample-level analysis. The first, which we refer to as exploratory analysis, is to divide the samples into groups on the basis of their cellular and molecular properties. The idea of de novo grouping has seen powerful applications in clinical studies that use functional genomics to enable more precise prognoses and treatment planning10,11. As a prominent example, pan-cancer analysis with functional genomics has revealed that, in many cases, people with cancer are more effectively classified using their molecular data, rather than histopathology12. The second task is to conduct comparative analysis, that is to identify cellular and molecular features that differ between predefined groups of samples (for example, cases versus controls). In bulk-level studies, differential expression (DE) has typically been used to detect gene expression programs that are associated with conditions of interest13. The advent of single-cell genomics also popularized differential abundance (DA) as another form of comparison to discover cell states that are disproportionately abundant in a given group of samples14.
Current approaches for these two closely related problems suffer from limitations that preclude them from taking full advantage of the resolution afforded by single-cell genomics. Starting with exploratory analysis, a common approach for quantifying the distances between samples is to first organize the cells into groups (representing types or states) and then evaluate the differences in the frequency of each group5,7,15,16,17. This approach, however, might oversimplify the task by reducing the amount of available rich information about each sample. Furthermore, it hinges on the effective clustering of the cells (to represent distinct cell states), which is often complicated by the need for high resolution, to distinguish between closely related states and harmonize samples or datasets. Finally, this approach can miss critical effects that might manifest in only particular subsets of cells (which we later demonstrate, using cohorts of people with irritable bowel disease (IBD) or COVID-19). Similar issues also emerge in comparative analyses. Most current applications of DE and DA rely on a priori clustering of cells. It is possible, however, that DE programs span few or parts of the a-priori-defined cell subsets and thus are less likely to be detected. Similarly, differentially abundant subpopulations might not clearly correspond to any annotated subset, again limiting the ability to detect them. Finally, even with access to high-quality cell annotation, comparative analyses of different partitions (for example, comparisons between sexes or age groups) might be best reflected by different cell-clustering schemes18.
To mitigate these issues, a recent line of work focused on quantifying DE or DA without relying on predefined cell clusters19,20,21. These methods typically embed cells into a low-dimensional space and then consider small neighborhoods in that space to identify ‘local’ DE or DA effects. A caveat of this approach, however, is that it does not account for the uncertainty that embeddings might have (for example, as inferred with variational autoencoders (VAEs)22), which can be substantial23. Another line of work uses VAEs to learn the effect of sample covariates on the latent embedding of cells24,25,26. The primary limitations of this approach are that it assumes the evaluated effects are constant, meaning they are identical for all cells irrespective of their state, and that they do not account for the uncertainty in estimating these effects.
To address these challenges, we introduce MrVI, a probabilistic framework for large-scale (multi-sample) single-cell genomics. For exploratory analysis, MrVI identifies sample groups without requiring a priori cell clustering. Instead, it allows for different sample groupings to be conferred by different cell subsets that are detected automatically. For comparative analysis, MrVI enables both DE and DA in an annotation-free manner and at high resolution, while accounting for uncertainty and controlling for undesired covariates, such as experimental batch. The notion at the basis of MrVI is that of counterfactual analysis, which aims to infer what would the gene expression profile of a cell be had it come from a certain sample. This approach provides a principled methodology for estimating the effects of sample-level covariates on gene expression at the level of an individual cell. It relies on a hierarchical deep generative model architecture, powered by modern techniques in deep learning, such as cross-attention, to model the effects of sample covariates while providing state-of-the-art performance in sample-integration quality. On the software side, MrVI leverages optimization procedures included in scvi-tools27, allowing it to scale to multi-sample studies with millions of cells.
In this work, we demonstrate that MrVI compares favorably to common approaches for integration, exploratory and comparative analyses and then showcase its utility in several multi-sample studies. In a PBMC dataset from a COVID-19 study, MrVI identifies a monocyte-specific response to the disease that more naive approaches cannot directly identify. In a dataset of drug-perturbation screens, MrVI reveals both expected and non-trivial relationships between compounds. Finally, using MrVI to study a cohort of people with IBD, we find a previously unappreciated subset of pericytes with strong transcriptional changes in people with stenosis.
Results
Multi-resolution variational inference
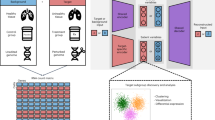
MrVI is a hierarchical Bayesian model for integrative, exploratory and comparative analysis of single-cell RNA-sequencing data from multiple samples (for example, corresponding to human participants) or experimental conditions (for example, perturbations in a screen; Fig. 1a). The model utilizes two levels of hierarchy to distinguish between two types of sample-level covariates. The first covariate type captures properties in either exploratory or comparative settings—we refer to these as target covariates.

a, We consider multi-batch, multi-sample experimental designs. In the canonical case, we gather single-cell measurements from several samples, which are collected across several batches. In this case, the relevant nuisance covariate is the batch. b, Left, MrVI model illustration. Right, graphical model plate diagram. MrVI relies on two cell representations, u and z. A sample-unaware cell representation (u) captures shared type information (colored by cell type in the diagram). From this quantity and the sample-of-origin of the cell, we construct a sample-aware representation (z) of the cell. Last, we model gene expression as a function of this latent variable and of observed nuisance factors. Each point in the diagram corresponds to an individual cell. c,d, Use cases of MrVI for exploratory and comparative analyses. c, For exploratory analysis, MrVI computes local sample stratifications. MrVI can compute counterfactual representations, characterizing what would have been the representation of a cell had it originated from a different sample. By computing the distances between counterfactual representations of all samples, MrVI can identify sample-level effects on cell states. d, For comparative analysis, MrVI quantifies differences in abundance across cell states (top right), and identifies sample metadata effects on gene expressions (bottom). Both the sample stratification and differential expression procedures use counterfactual z representations to compare local sample effects. The differential abundance procedure involves an approximation of the posterior density for each sample in the u latent space.
Typically, an identifier for each sample (for example, human donor ID or experimental perturbation) is a natural choice for the target covariate to be provided as input to MrVI, because it is entirely nested in other sample-level target attributes (for example, treatment type), thus enabling their analysis. The second covariate type is considered ‘nuisance,’ and typically corresponds to technical factors (sample processing site, library-preparation technology or the study of origin in cross-studies).
In MrVI, each cell (n) is associated with two low-dimensional latent variables, un and zn (Fig. 1b): un is designed to capture the variation between cell states while being disentangled from sample covariates; zn, reflects the variation between cell states, in addition to the variation induced by target covariates, while remaining unaffected by the nuisance covariates. Finally, we model the observed gene expression (xn) as samples from negative binomial distributions whose parameters are predicted by decoding zn conditioned on nuisance covariates.
MrVI employs a mixture of Gaussians as a prior for un instead of a uni-modal Gaussian. We demonstrate that this more versatile prior provides state-of-the-art performance in the integration of large datasets and in facilitating annotations of cell types and states. zn is learned as a function of the respective un and the sample ID, sn (Methods). We used neural networks for all mapping functions in the model. The parameters characterizing these functions are learned through maximization of the evidence lower bound (Methods)28.
The trained model performs two types of analyses at single-cell resolution—exploratory (de novo grouping of samples) and comparative (evaluating the effects of target covariates). For exploratory analysis, MrVI computes a sample-by-sample distance matrix, or sample distance matrix in short, for each cell n by evaluating how the sample of origin (sn) affects the representation of this cell in z space (Fig. 1c). To this end, for each cell n, we compute \(p({z}_{n}| {u}_{n},{s}^{{\prime} })\), its hypothetical state had it originated from sample \({s}^{{\prime} }\ne {s}_{n}\). We then define the distance between each pair of samples on cell n as the Euclidean distance between their respective hypothetical states. Then, hierarchical clustering can be used over the sample distance matrices for each cell to highlight the target covariates most likely to explain the major axes of sample-level variation. This analysis helps capture, in an annotation-free manner, cellular populations that are influenced distinctly by target covariates (for example, disease or tissue of origin).
In comparative analysis, MrVI identifies both DE and DA at single-cell resolution (Fig. 1d) using counterfactuals. Consider the case of differential expression between two sets of samples (S1, S2). To evaluate the group-level effects in cell n, we evaluate the extent to which the expectation of \(p({z}_{n}| {u}_{n},{s}^{{\prime} })\) depends on whether \({s}^{{\prime} }\) is in S1 or S2 using a linear model. We then use the decoder network (that is, mapping from z to x) to detect which genes are affected and evaluate their effect size (fold change). Contrary to traditional DE methods, MrVI does not require pre-existing grouping of the data upstream of model fitting. Meanwhile, for local differential abundance, we estimate the posteriors (\(p({u}_{n}| {s}^{{\prime} })\)) and compare the aggregate values of samples \({s}^{{\prime} }\) in S1 versus S2. An in-depth description of MrVI and its post-training analysis procedures is provided in the Methods.
Retrieving known sample effects on a semi-synthetic dataset
We used a semi-synthetic dataset to evaluate how accurately MrVI captures differences between samples (through exploratory and comparative analysis) when different cell subsets are influenced by different sample-level effects. Taking a published dataset of 68,000 peripheral blood mononuclear cells (PBMCs)29 profiled with 10x, consisting of 3,000 highly variable genes and five main cell clusters, which we refer to as subsets A–E. We assigned each cell in this dataset to 1 of 32 synthetic study subjects. These study subjects are characterized by two distinct sample-level covariates. Our strategy for assigning cells to the simulated subjects varied between the cell subsets to simulate different covariate effects. For subset A, the assignment of cells resulted in DE across categories of covariate 1, reflecting a hierarchical grouping of the samples. For subsets B and C, our cell assignment reflected DA across categories of covariate 2 (Fig. 2a). Cells in the remaining subsets were randomly assigned to samples and hence did not contain any DE or DA effects (Methods).

a, Experimental design. We created a semi-synthetic dataset with 5 subsets of cells and 32 study subjects (subj.), containing sample-specific differential-expression (exp.) and differential-abundance effects. In cell subset A, cells have differences in gene expression, on the basis of the value of a study subject covariate, covariate 1. These DE effects stratify synthetic samples according to a known hierarchy. In subsets B and C, cells have differences in abundance between study subjects, on the basis of the value of a second study subject covariate, covariate 2 (four categories (cat.) in total). Cells in subset B or C were over- or under-sampled, respectively, according to fixed rates in each category, such that the sum of cells from B and C remained constant. Stars indicate categories with strong resampling. There are no DE or DA effects across study subjects in other cell subsets. b, Minimum distortion embeddings (MDEs) of MrVI u and z latent spaces, colored by cell subset assignments and covariate 1 categories. c, MrVI’s distance matrices aggregated over cell subsets A and B. d,e, DA analysis using MrVI and Milo for the comparison of synthetic samples based on covariate 2 categories (categories with strong DA in cell subsets B and C versus rest (see a)). d, A u latent space MDE, colored by log density ratios comparing the subset population (star) with the remaining (rest) population (Equation (4)). Enr., enriched. e, Precision–recall curves with areas under the curve (AUCs, higher is better) for identifying DA cells. Cells in subset B or C are true positives; other cells are true negatives. We used the absolute value of the log density ratio for MrVI and the absolute value of the LFC produced by Milo as scores to estimate precision–recall. f,g, DE analysis using MrVI and miloDE comparing group 1 of the simulation (4 blue figures) against all other samples. We inferred which cells showed DE effects for the comparison of synthetic samples based their covariate 1 assignment (blue versus rest). f, u latent space MDE colored by the squared norm of βn, appearing in Equation (3), which quantifies the overall sample covariate effect on gene expression. g, Comparison of miloDE and MrVI LFCs versus DESeq2 reference, reporting Pearson’s r for each method.
We applied MrVI using the simulated subject identifiers as the modeled target covariate (sn) and leaving the nuisance covariate (bn) empty. The resulting u space clearly reflected the differences between the cell subsets (Fig. 2b). In the z space, we observed distinct subject-specific effects in cells of subset A, whereas cells in the remaining clusters were mixed, aligning with the expectations that subset A alone contained DE effects. For exploratory analysis, we used the mapping from u to z to estimate sample distances for each cell (Fig. 2c). In cell subset A, the sample distance matrix (averaged over cells) produced a hierarchical structure similar to the simulated (ground truth) dendrogram. As expected, MrVI estimated much smaller distances between samples when considering the other cell subsets, with no discernible structure. We compared this result with the standard approach for stratifying subjects using clustering obtained either from PCA or scVI (Methods). The resulting compositional analyses were less effective in capturing sample stratification in subset A (Supplementary Fig. 1a,c) and introduced non-negligible distances in subsets in which no differences were expected (Supplementary Fig. 1b).
For DA analysis, we partitioned the subjects into two groups, according to covariate 2 (presence or absence of a star in Figure 2a). We used the estimated posteriors (p(u∣s)) around each cell to evaluate the extent to which its state was over-represented in one group of study subjects versus another. The resulting log ratios accurately reflected the DA effects that were simulated in cell subsets B and C (Fig. 2d). Furthermore, the inferred ratios significantly diverged from zero in only subsets B and C (Supplementary Fig. 1d). We compared MrVI with Milo19, a popular framework for DA, and found that MrVI more accurately identified DA effects and associated them with the correct cell subsets (Fig. 2e and Methods).
For DE analysis, we compared the subjects in one category of covariate 1 (blue in Fig. 2a) with all other subjects. In this comparison, only cell subset A was expected to contain DE effects. We used the estimated posteriors (p(z∣u, s)) around each cell to evaluate the extent to which its gene expression profile depends on its sample-of-origin category, using the linear model (in latent space) to obtain effect sizes (Methods).
These quantities reached much higher values in subset A cells than in cells in other subsets (Fig. 2f), indicating that MrVI captured the particular groups of cells exhibiting DE effects. Next, we evaluated each gene’s effect size (log fold change; LFC) in each cell belonging to subset A using the MrVI model. We compared these values with those obtained through pseudo-bulk DE analysis of subset A (representing an annotation-dependent analysis in the ‘perfect’ scenario, in which the annotations completely align with the DE signal). The results from these strategies were highly correlated, with a substantial improvement over miloDE—a recent cluster-free method for DE analysis (Fig. 2g).
These results demonstrate that MrVI can identify different sample groupings for different cell subsets without requiring an a priori annotation of cell states. Similarly, it can accurately retrieve shifts in cell-state composition (DA) and gene expression (DE) and identify the respective cellular populations.
Highlighting variation in myeloid responses of people with COVID-19
We next used MrVI to analyze 419,000 PBMCs obtained from a cohort of people with COVID-19 and healthy controls7. We used the sample identifier, corresponding to unique study participants, as our modeled target covariate (s). As anticipated, the resulting u space is not affected by the sample of origin, instead showing marked mixing between study participants (Fig. 3a). At the same time, the u space clearly stratified the cells into immune subsets in a manner consistent with their annotation in the original study. Considering a standard evaluation of integration performance30, we found that the MrVI u space embedding outperformed PCA and scVI in terms of mixing the samples while retaining their biological signal30 (Supplementary Fig. 2). However, a model should capture the effects of viral infection. The two-level structure of MrVI allowed us to derive both a representation that is cell-type centric (u) and one that is affected by the respective sample (z). Indeed, the z space showed clear sample-specific variation, separating COVID-19-positive individuals from the control population inside each cell type (Fig. 3b).

a,b, MDEs of u and z latent spaces in MrVI, computed on the full dataset and colored by the original cell-type annotations and COVID-19 status. pDC, plasmacytoid DC. c, Sankey plot mapping cell-type annotations to clusters obtained by clustering cell-specific distance matrices using the Leiden algorithm. This clustering identified three cell subpopulations, A, B and C. Cluster A contained monocytes and DCs, cluster B contained T cells and NK cells and cluster C contained B cells. Cell-type or cluster pairs with less than 1% of the total cells are not shown. d, Sample distance matrices averaged over cells from two of the three subpopulations in c. For each matrix, we computed the associated affinity dendrogram between samples obtained through hierarchical clustering and colored each row (sample) according to participant age, DSS, infection status and the most severe stage of disease that a participant has experienced. e, Differential abundance analysis using MrVI log density ratios for the myeloid cells identified as cluster A in c. Left, comparison of COVID-19-positive individuals with healthy controls. Right, comparison between COVID-19-positive individuals with high or low DSS. f, Differential expression analysis using MrVI between COVID-19-positive individuals with high or low DSS. MrVI identified three DE modules of genes. Each plot shows the activity of the module in the u latent space. Displayed are the LFCs averaged over all genes in the module. In these figures, the individuals with low DSS and those with high DSS, respectively, correspond to donor clusters 1 and 3 in d. Upreg., upregulated; expr., expression.
We used MrVI to answer two fundamental questions: how do samples in this cohort stratify into groups? Do they stratify differently when considering different immune populations? To address these topics, we used counterfactual embeddings to estimate a sample distance matrix for each cell (n). We then clustered the cells according to these values, thus detecting groups of cells inducing similar sample stratifications (Fig. 3c). This analysis produced three groups of cells, one containing T cells and natural killer (NK) cells, another consisting primarily of monocytes along with a smaller population of dendritic cells (DCs), and a third containing B cells.
The resulting distance matrices (averaged across all cells in each respective group) separated groups of people with disease and controls, indicating that MrVI can identify clinically relevant groups (Fig. 3d and Supplementary Fig. 3). However, the distance matrix conferred by monocytes and DCs highlighted an additional stratification of the study participants. In this cluster, the COVID-19 group was further stratified into two groups, corresponding to groups 1 and 3 in Figure 3d. Group 1 was enriched in individuals for whom the number of days since first symptoms (DSS) was low, whereas individuals in group 3 showed longer duration of symptoms (Extended Data Figure 1; Mann–Whitney U test, P < 0.05). The association with monocyte activity and the time elapsed since infection has been established31. MrVI identified this association without prior knowledge of the DSS or any other information about the participants.
To interpret this data-driven stratification of people with COVID-19 and its association with monocytes, we performed two analyses. We first performed a DA analysis of the myeloid population, comparing those in the COVID-19 and control groups. We found a marked decrease in non-classical CD16+ monocytes and DCs (Fig. 3e and Supplementary Fig. 4a) in people with the disease. The comparison of the two COVID-19 groups similarly showed a shift toward non-classical monocytes in the group with higher DSS (Fig. 3e and Supplementary Fig. 4b). These results are consistent with independent studies31, which reported that CD14+ monocytes are highly pro-inflammatory and contribute to the cytokine release in early COVID-19, thereby contributing to symptoms.
Next, we applied our DE analysis to compare the two COVID-19 groups. Using MrVI counterfactuals, we estimated the respective LFC for each gene in every myeloid cell. We then clustered the genes on the basis of their estimated LFC profiles (Methods). This analysis uncovered three modules, each containing genes with a similar DE pattern, implicating different subsets of myeloid cells (Fig. 3f and Supplementary Fig. 5). The first module, upregulated in the group with higher DSS, was enriched in genes identified in myeloid cells of healthy individuals (compared with those with the disease), again supporting the notion of a return to baseline with long-standing infection. Specifically, we see a lower CSF3R expression in the recently infected individuals, aligning with less-mature monocytes that are released earlier from bone marrow during infection32. Similarly, we found that, early in the infection, the number of MHC-II-expressing monocytes declines but later returns to normal levels31. This accounts for the observed elevation in LGALS2 and HLA-DR2, both linked to MHC-II. The second module, over-expressed in individuals with lower DSS, is enriched in interferon-related genes. This module includes GBP1 and IFITM3, interferon-response genes, and IFI27, reported as an early predictor of COVID-19 severity33. These results agree with strong interferon signaling during early infection, especially in myeloid cells31. The third module, over-expressed by the higher DSS group, contained TNF and NFKBIZ. It has been demonstrated that tumor necrosis factor (TNF) release is reduced during acute COVID-19, whereas NFKBIZ expression is drastically reduced during acute infection34. Our analysis suggests that both molecules are markers of acute infection more so than mortality.
Grouping molecular effects on expression from perturbations
To demonstrate the flexibility of the target covariate used by MrVI, we analyzed a chemical perturbation screen with a single-cell RNA-seq readout generated with the sci-Plex assay6. The sci-RNA-seq3 dataset includes three cell lines, 188 small-molecule drugs and vehicle controls. Each small molecule was delivered at four doses, and the entire study was conducted with two biological replicates. In this assay, each cell receives a single perturbation (or negative control vehicle) that can be identified in addition to its transcriptome. MrVI can serve several fundamental analyses for this type of study, namely integrating all replicates into a shared embedding, stratifying the screened compounds into groups with similar effects, mapping these effects at the gene-expression level and mapping cell-state composition.
To achieve this, we used the concatenation of the drug name and the dose level as the target covariate modeled by MrVI, resulting in 752 ‘samples’ per cell line. Because the study was conducted with 96-well plates, with each plate containing one of two biological replicates, we chose the plate identifier as our nuisance covariate. As found in the original study, many drug–dose combinations had minimal effect on transcription across the assayed cell lines6. Consequently, we applied a simple filter, retaining only drugs that had a minimal number of DE genes with at least one concentration and in at least one cell line (Methods). This resulted in 368 perturbation samples (92 drugs with four concentrations each) that we used to train MrVI (Methods). Here, we focus on the epithelial A549 lung adenocarcinoma line and provide the results from the other two cell lines in the Supplementary Information.
The resulting u space (Fig. 4a) appears as a single cluster with no apparent sub-clusters specific to a given class of drugs, indicating a successful integration that reflects the drug-independent states of the cells. We rather observed that positioning in the u space carries information about the cell cycle. With respect to the perturbation-affected (z) space (Fig. 4b), we observed several sub-clusters of cells originating from distinct classes of drugs. In particular, populations of cells treated with HDAC inhibitors (expected to target epigenetic regulation) and trametinib (to block MEK-mediated tyrosine kinase signaling) formed clear clusters, highlighting their distinct drug-induced shifts in gene expression. The distinction of HDAC inhibitors is in agreement with the original study, in which the authors additionally identified acetyl-CoA deprivation as a common mechanism for this drug class, captured by drug-induced shifts in gene expression. The response to trametinib is also expected to greatly impact the Ras-driven A549 cells because the drug inhibits the downstream MEK pathway6.

MrVI was fit over 92 drugs each at four doses that passed our simple DE-gene filter. a,b, MDEs of the u and z latent spaces, colored by the pathway of the drug used to treat each cell (left) and the cell-cycle stage of each cell (right). For the MDEs colored by pathway, only the top 20% of samples, on the basis of distance from the vehicle, are shown in full opacity. c, PCA of sample distance matrices. Left, scatterplot of all local sample distance matrices projected onto the top two principal components, colored by cell-cycle stage (displays no visual subclusters). Right, barplot of the proportion of variance explained against the number of principal components. d, Comparison of MrVI against the benchmark methods, on the basis of performance metrics assessing alignment with prior knowledge. Each bar represents the metric for one model fit, except for ‘Random,’ which reports the 95% confidence interval over 100 permutations of the inferred distance matrix from MrVI. Left, average percentile of intra-drug distances, measuring how much closer samples with the same drug and different doses are to each other relative to other samples (lower is better). Right, silhouette score of sample clusters with similarities inferred from DEG sets in the connectivity map dataset, assessing cluster consistency (higher is better). e, Hierarchically clustered sample distance matrix. Rows are annotated by the pathway, dose and cluster of each sample (clusters inferred from the distance matrix). f, Heatmap of gene set enrichment analysis (GSEA) scores for the Human MSigDB Hallmark gene set collection for DE genes identified for each cluster in e. Each tile’s upper-right and bottom-left triangles, respectively, represent scores for the set of upregulated (Upreg.) and downregulated (Downreg.) DE genes. For e and f, the analysis is performed over the top 20% of drug–dose combinations (74/368), on the basis of their distance from the vehicle (see Extended Data Fig. 2 for the full matrix and Supplementary Fig. 7 for the matrix here with drug–dose labels).
For a more quantitative comparison, we computed MrVI sample distance matrices. We found that the cells in this cell-line-based assay are homogeneous in terms of the distance matrices that they induce (Fig. 4c), with no evident subclusters in PC space. By contrast, other datasets analyzed here explored primary cells with diverse cell types, featuring distinct distance matrices (Figs. 3 and 5). Therefore, we operated on one sample distance matrix, averaging over all cells.

a, Uniform manifold approximation and projection (UMAP) embedding of u latent space, colored by different sample-level covariates. From left to right, coarse labels, identified by us to stratify cell types for unguided analysis (circled is the subset analyzed in b–e); the tissue-collection method highlights a bias for stromal cells in surgical specimens; the inferred effect size of inflamed versus non-inflamed and the inferred effect size of B2 disease behavior (highlighted are the small subpopulations of pericytes with the strongest effect (Supplementary Fig. 20). For these last two plots, this effect size corresponds to the squared norm of β from Equation (3); it characterizes the overall effect of the covariate on gene expression. b–e, Analysis of the cell population circled in a, using the same UMAP embeddings as in a. b, Cell-type labels provided in the original study. Displayed are fine annotations for stromal cells and coarse labels for the other cells. c, DA analysis of stromal cells. MrVI was used to compare the B2 disease phenotype and the B1 phenotype (red denotes higher in B2 disease behavior). Displayed is the log (density ratio) between B2 and B1 disease behavior. The number of cells of each type is reported in Supplementary Table 4. d, Raw expression of CDH11 inside all stromal cells. Values are library-size-normalized and log1p-transformed. Cells are sorted for display on the basis of higher expression. e, Inferred LFCs from MrVI for the comparison of B2 and B1 disease behavior (beh.) on the basis of multivariate analysis in MrVI, correcting for inflammation status, sex, tissue location and chemistry in the different cell types. The violin plots display the distribution of estimated LFCs per cell type. Hierarchical clustering was used to determine the order of cell types in the violin plots. f, UMAP of two genes, highlighting intra-cell-type DE variation. Cells are colored on the basis of the genes’ LFC for the comparison of B1 and B2 classifications.
To test whether the resulting distance matrix captures a priori known relationships between the samples, we formulated two performance metrics and compared MrVI with the two standard (composition-based) methods (Fig. 4d and Supplementary Information B.1). First, we used the transcriptomic-based Connectivity Map resource35, providing a measure of similarity between drugs, and compared these similarities with the distances we estimated using MrVI for the maximum tested dose (10,000 nM). Second, we evaluated the extent to which treatments with the same drug but at different concentrations tend to be more similar to each other than expected by chance. MrVI achieved better performance on both metrics—showing higher concordance with the Connectivity Map stratification, and lower distances between treatments with the same compound. These metrics were also used for fine-tuning the hyperparameters of the MrVI model (u and z dimensions). This strategy reflects real-world cases in which prior knowledge of the similarity between samples can be utilized for more effective modeling and downstream analysis (Supplementary Fig. 6).
We then analyzed a hierarchical clustering of the sample distance matrix (Fig. 4e and Supplementary Fig. 7). Drug-dose combinations that had little to no effect in the A549 context mostly clustered with the vehicle treatment (Extended Data Figure 2). In particular, a disproportionately high number of samples with low effect sizes also had low dosages, reflecting expected dose–response relationships. Of the remaining samples, those that were most distinct from the vehicle sample were organized into several clusters, each with a different effect on gene expression (Supplementary Figs. 8 and 9). Clusters 1, 3 and 4 consist mostly of HDAC inhibitors. These three clusters span a wide range of effect sizes that are correlated with the dosages. Cluster 1 contains the samples with the highest dosage levels and the largest effects, and cluster 3 consists of samples with lower dosages and weaker effects (Supplementary Fig. 8). These groupings highlight the ability of MrVI to uncover dose-dependent effects on gene expression that are apparent across multiple drugs in the HDAC inhibitor class. Clusters 2 and 8 corresponded to all doses of trametinib and YM155, respectively. In these cases, MrVI therefore suggests that the effects of these drugs were less dependent on the dose, at least when considering the range of concentrations tested.
MrVI also uncovered relationships between drugs, on the basis of their effects on transcription, that are supported by recent literature and the original sci-Plex study. For instance, cluster 8 includes rigosertib, which is labeled as a tyrosine kinase inhibitor but has been found to directly affect microtubule function36, as well as epothilone A and patupilone, two drugs that interfere with microtubule function. Moreover, MrVI revealed non-trivial similarities that were not captured in the original study. In cluster 5, two JAK2 inhibitors, fedratinib and TG101209, were grouped with JQ1, a drug labeled as a BRD inhibitor. Notably, recent work has shown that JQ1 inhibits the JAK–STAT signaling pathway in addition to being a BRD inhibitor, which supports the plausibility of this grouping37.
Finally, we investigated the clusters by performing Gene Set Enrichment Analysis (GSEA38) on the DE gene sets identified by MrVI (comparing each cluster of samples to the vehicle controls; Fig. 4f and Methods). As a reference, we used the hallmark collection of MSigDB39 that records sets of genes that contribute to major cellular processes. This analysis shed additional light on the effects of each cluster of drugs. Specifically, we found that clusters 1, 3 and 4 associate most strongly with downregulation in metabolic pathways, agreeing with the effect of HDAC inhibitors on carbon metabolism6. Furthermore, cluster 6 was enriched in the p53 pathway, consistent with the categorization of its respective drugs as cell-cycle regulators. Similarly, the effects of cluster 2 were enriched in genes downstream of KRAS signaling, in agreement with its categorization as targeting tyrosine kinases. We provide a heatmap of the LFCs for the top DE genes across all clusters in Supplementary Figure 10. On the basis of this analysis, we highlight that MrVI not only provides an interpretable grouping of clusters, but additionally helps highlight the genes underlying this grouping.
For the other two cell lines used in the sci-Plex experiment, the results of MrVI were consistent with known biology (Extended Data Figs. 3 and 4 and Supplementary Figs. 11–18)40,41.
Profiling stromal cell dynamics in Crohn’s disease stenosis
To provide another example of the applicability of MrVI to human cohorts, we utilized a recent study conducted in 46 people with Chron’s disease and 25 controls across 463,000 cells using single-cell RNA-sequencing42. The dataset includes metadata describing the anatomical location of sampling (in terms of tissue, colon or ileum; in terms of the tissue layer, lamina propria or epithelial), the method of extraction (surgical or biopsy) and sample-preparation detail (10X chemistry). It also includes information on individual study participants, such as disease state and the presence of stenosis in their history. We used these metadata to evaluate the ability of MrVI to recognize meaningful subgroups of people with Chron’s and to highlight cell populations that are affected by stenosis.
We trained MrVI to integrate all the samples in this dataset, using the sample identifier as the modeled target covariate and the combination of library preparation protocol and tissue layers (lamina propria and mucosa) as the nuisance covariate. Comparing the resulting u space with the embedding obtained with scVI, we found that the default settings of MrVI yielded better mixing between the study participants but had slightly lower performance in terms of distinguishing between cell states (using cell annotations assigned in the original study; Extended Data Fig. 5). Indeed, integration is challenging in this dataset owing to significant differences in cell-type composition in the colon and in the ileum, as well as between the mucosal and lamina propria layers. To enhance the alignment of u with known cell states, we developed a variant of MrVI that incorporates cell-type labels (Methods). This variant exhibited overall improved performance compared with scVI, managing sample mixing while preserving cell-type information (Extended Data Figure 5).
We next used MrVI to explore how the different samples are stratified, and how these strata change between cell types. Using a coarse definition of cell types (Fig. 5a and Methods), we again find that different types are associated with different groupings of people with the disease. For instance, considering a subset of immature enterocytes (referred to herein as enterocytes-stem), the sample distance matrix clustered solely by their tissue-of-origin (colon or ileum) (Supplementary Fig. 19). We applied MrVI’s DE function on the enterocytes-stem subset to investigate the differences between these clusters. In line with their biological functions, we observed higher expression of AQP8, CA2 and CA8 in the colon, all of which encode proteins that absorb water, and higher FABP6 and FABP2 expression in the ileum, which encode proteins that absorb fatty acids. Considering that a population of mature enterocytes provides a slightly different view, highlighting a specific cluster of eight people with Chron’s that are distinguished from all others with the disease and controls. Using MrVI DE to compare mature enterocytes in this cluster, which mainly contained colon samples, with other colon samples, we detected an upregulation of genes encoding mucins (MUC1, MUC2, MUC12), which is a well-described pattern in Crohn’s disease43, and upregulation of CXCL1 and CXCL3, which encode chemokines that attract neutrophils44. The expression of these chemokines was associated with stimulation of epithelial cells with IL22 and IL17A, key cytokines in Crohn’s disease. Furthermore, neutrophil infiltration is a key feature of inflamed gut regions45. Depending on the cell subset under consideration, the exploratory analysis of MrVI reflected known differences between the tissues sampled (here, ileum versus colon) and revealed differences in a subpopulation of people with the disease (non-inflamed versus inflamed).
Next, we used MrVI for comparative analysis with respect to a known covariate. We considered the distinction between the two most common complications of Crohn’s disease: stenosis (Vienna classification B2, 11 participants) and fistula or abscesses (penetrating; Vienna classification B3, 7 participants). The remaining are healthy controls or individuals without either complication (Vienna classification B1). Finding reliable biomarkers to distinguish between individuals experiencing the two types of complications is critical, as they might require different treatment strategies. Using our multivariate DE procedure, we studied the presence of stenosis and the inflammation status (inflamed versus non-inflamed) of individual cells, while accounting for the effects of nuisance covariates such as biological sex and tissue location (Methods). We excluded surgical samples from this analysis owing to the marked differences in cell-type composition compared with biopsies (which are the source of most cells in the dataset; Fig. 5a and Methods). Inflammation status had a marked effect on several cell lineages and a strong effect on the stromal compartment. As expected, the presence of B2 disease had a more mild effect, mostly restricted to a few stromal subsets, with its highest impact in a small subset of pericytes (Fig. 5a, Supplementary Fig. 20 and Extended Data Fig. 6b). Therefore, the remainder of the analysis focused on stromal populations consisting of fibroblasts, pericytes, glial cells and endothelial cells (Fig. 5b and Supplementary Fig. 21).
We first compared B2 with B1 samples using DA analysis, controlling for inflammation status and other covariates (Methods). We find a decrease in the abundance of several endothelial populations in B2 samples (for example, lymphatic and LTC4S+ endothelial cells) and an increase in fibroblast populations (for example, ADAMDEC1+; Fig. 5b,c and Extended Data Fig. 7b). This result is in accordance with the prevalence of microvascular rarefaction (that is, loss of endothelial cells) in tissue fibrosis46. Using the same settings for DE, we find that in individuals classified as B2, CDH11, a biomarker of stenosis47, as well as classical markers of tissue fibrosis (ADAMDEC1 and COL1A1) and activation (S100A6, MT2A, and JUN), were upregulated in a subset of HIGD1B+STEAP4+ pericytes (Fig. 5d–f and Extended Data Fig. 6d). Although these genes are upregulated in B2 disease by other stromal subsets, we find several genes that are affected uniquely in cells of the HIGD1B+STEAP4+ pericyte subset. This includes LUM, which is upregulated after fibroblast stimulation in lung fibrosis and promotes fibrocyte differentiation48, and PDGFRB and TGFBI, which are strongly upregulated in lung fibrosis and whose pharmacological inhibition reduces fibrosis20,49. Furthermore, LUM and TGFBI have been reported to be upregulated in the chronic phase of a mouse model of colitis50, which is associated with marked intestinal fibrosis. Notably, although MrVI bases its DE estimations on its generative model, targeted analysis of the same molecules using the raw data shows consistent results (Extended Data Fig. 7a). Together, this analysis demonstrates the potential of MrVI for delineating a population of cells that is associated with a disease phenotype and could facilitate a more nuanced discovery of markers for diagnosis and treatment.
MrVI additionally predicts marked upregulation of PDGFRB by CD36+ endothelial cells in B2 samples (Fig. 5e,f). This is unexpected because PDGFRB is a common marker of pericytes and is not normally expressed by endothelial cells. We further characterized gene expression in the CD36+ subset and found coexpression of endothelial markers (like PLVAP, VAMP5, VWA1) and pericyte markers (like NOTCH3, RGS5 and MYL9) (Extended Data Figs. 6d and 7b). We also found upregulation of markers of tissue fibrosis, such as COL1A1 and TGFBI, in this subset (Extended Data Fig. 6e). Therefore, these results highlight a cell population with a mixed phenotype between the endothelial and pericyte lineages, which upregulates markers of tissue fibrosis in B2 samples. This hints at the endothelial-to-mesenchymal transition in IBD and the presence of a pericyte-like state in the gut endothelium of B2 disease. This transition has been described as occurring in human IBD51. However, the phenomenon was not explored in the original study of this cohort, and has not to date been studied with single-cell genomics51.
Discussion
In this paper, we introduce MrVI, a comprehensive solution for large-scale (multi-sample) single-cell RNA-seq studies. MrVI provides a unified probabilistic framework for integration of samples, sample stratification and analysis of the effects of sample covariates at both the cell-subset and gene levels. Based on a hierarchical latent variable model and counterfactual predictions, MrVI addresses these tasks while accounting for nuisance sources of variation, without requiring cell-type annotations.
The latter point is of particular importance owing to the difficulty of defining cluster boundaries and their resolution. For instance, ref. 10 categorized the human brain into 17 cell types for studying autism, whereas ref. 52 identified 3,313 clusters to characterize cellular heterogeneity in the same tissue. Both strategies proved useful, and it is therefore not generally clear which resolution is appropriate for a given analysis. Agnostic to such clustering strategies, MrVI facilitates a ‘bottom up’ approach that divides the cells into groups in a manner that reflects the task at hand. Specifically, by estimating sample distance matrices around each cell, MrVI aggregates cells into subsets that confer similar groupings of samples. Similarly, estimating DE or DA effects in every cell allows for the aggregation of genes or cells in a way that reflects a coherent response to the covariate of interest. For ease of interpretation, these aggregations can also make use of cell annotations into subsets (by averaging the cell-wise DE or DA effects), as long as the cells in a subset are consistently affected (which was the case in many of our analyses).
MrVI’s architecture uses multi-head attention to model heterogeneous covariate effects, a distinction from traditional models that typically rely on multi-layered perceptrons. We validated this design using ablation studies motivating these choices (Supplementary Fig. 22). We also guided the selection of default parameters that are relevant for most use cases through hyperparameter sensitivity studies (Supplementary Fig. 22, Supplementary Tables 2 and 3 and Supplementary Note B).
We demonstrated MrVI performance in a few case studies. Considering a COVID-19 cohort, MrVI identified clinically relevant groupings of people with the disease and highlighted subsets of myeloid cells in which these groupings manifested. Post hoc analysis of the resulting strata revealed a marked agreement with the elapsed time since infection—information unavailable to the algorithm. Notably, MrVI’s grouping did not perfectly mirror the infection timelines. This observation underscores MrVI’s potential to produce data-driven sample strata that might not be trivially obtained from the recorded metadata alone and instead could lead to different diagnoses or identification of new disease subtypes12. MrVI is particularly relevant for studies in which samples are collected from numerous individuals and across different anatomical locations or experimental protocols. We demonstrated this using an IBD study, in which MrVI effectively integrated samples from diverse tissue locations and highlighted changes associated with stenosis. Beyond clinical or cross-studies, MrVI applies to any discrete cell-level meta-data by designating it as the target covariate. We demonstrated this using a perturbation screen with the sci-Plex assay, in which each cell is associated with a particular perturbagen, facilitating de novo identification of compound groups and characterization of their effects.
When considering patient cohorts, we applied MrVI using the sample identifier as our modeled target covariate (s). MrVI’s ability to stratify samples without relying on explicit covariate information makes it robust to scenarios in which metadata might be incomplete or inaccurate. There are, however, scenarios in which it is useful to explicitly model other target or nuisance covariates, which could constitute natural extensions of MrVI.
Another natural extension of MrVI is to handle information from other measurement modalities, both separately and in parallel to RNA expression. This extension could pinpoint, for instance, different strata (and their inducing cell subsets) when considering chromatin properties versus RNA53.
MrVI is implemented using state-of-the-art software tools for deep probabilistic modeling and can thus scale to multi-sample studies with millions of cells. Beyond that, the expected increase in scale and complexity of single-cell omics raise new challenges and opportunities for which MrVI can provide a powerful framework for analysis and a solid foundation for further developments.
Methods
The MrVI model
Generative model overview
We consider two-stage scRNA-seq experimental designs in which cells are collected from multiple samples (Fig. 1a). Each sample is associated with target covariates (for example, treated versus untreated, donor or specimen age and sex) or nuisance covariates (for example, the sample collection site or the study ID in cross-study analyses).
Typically, multiple target covariates can induce variation in expression across samples, but it is unknown which of these can affect cells and by what mechanism. For instance, in drug-response studies, both the type of administered drug and its dosage are crucial to assessing drug impact on cell states, but the nature of the interaction between these two factors might not be known. In disease studies, cases can induce specific shifts in gene expression in specific donor subpopulations that might not be fully encoded in the available metadata.
Instead of attempting to model the effects of these covariates directly, we adopt an approach that initially requires only knowledge of sample IDs s ∈ {1, …, S} and the nuisance covariates as b ∈ {1, …, B}. This strategy allows us, at a later stage, to highlight which target covariates drive sample variations of interest. The resulting gene expression profiles are denoted as {x1, …, xN}, in which \({x}_{n}\in {{\mathbb{N}}}^{G}\) is the vector of RNA transcript counts for cell n over the G observed genes. We denote the count for cell n and gene g as xng. For any cell (n), sn is the sample ID (for example, the donor from which cell n originates) and bn is the nuisance covariate.
In cases with multiple nuisance covariates, we recommend using the covariate with the coarsest resolution that is still nested in any covariates expected to confound the analysis. This might require concatenating multiple nuisance covariates (that is, the concatenation of the study ID with the batch ID used in each study as bn).
Isolating sample-specific effects on cell states with MrVI
The generative model of MrVI writes as:
Here, un and zn are the latent (unobserved) representations of cell n, both of dimension L. Azh is a matrix of dimension G × L, and γzh is a bias vector of dimension G. \({g}_{\theta }^{u\to z},{g}_{\theta }^{z\to h}\) are multi-head attention layers, with disjoint sets of parameters both denoted as θ for brevity. The size factor ln is fixed as the total sum of counts of cell n, hng denotes the normalized gene expression levels and rng ≥ 0 denotes the inverse dispersion of the distribution for cell n and gene g. μi, Σi and πi, respectively, denote the mean, covariance matrix, and weight of component i ≤ K, where K represents the total number of mixture components. More details about this prior are provided in ‘Additional model details’. All these parameters, other than ln, are learned during training.
A priori, un captures broad variations assumed to characterize cell types and more granular cell states, but is disentangled from both target and nuisance covariates. As such, un harmonizes cells from all samples into a shared latent space. We assume a mixture of Gaussians (MoG) prior or an unimodal Gaussian prior (K = 1) on un, depending on the application and available prior knowledge about cell-state variation. When we expect cells to belong to one of several groups, a MoG prior could be more appropriate than a unimodal Gaussian prior, to avoid posterior collapse and prior overregularization, two issues with variational inference that have been reported in the field54,55.
When reliable cell-type annotations are available, MrVI can also rely on a prior that is weakly informed about cell-type annotations. We found the cell-type-supervised model separated existing cell-type annotations much better than did its unsupervised counterpart (Supplementary Fig. 21). More details about the prior of un are given in ‘Additional model details’.
zn is an augmented representation of the cell, that is aware of sample effects but is disentangled from other nuisance covariates. This latent variable is constrained to be close to un by a term in the objective function that penalizes the L2 distance between zn and un.
These assumptions on un and zn hold a priori, but not necessarily a posteriori. For instance, the posterior distribution of un could exhibit conditional dependence on sample-specific factors, allowing MrVI to capture compositional differences across samples while harmonizing cells into a shared latent space.
Given that zn is expected to capture more variability in cells than is un, allowing it to lie in a higher-dimensional space is natural. Additionally, a low-dimensional bottleneck on un could improve sample harmonization. In such a case, we allow zn to take a higher dimension than un by modeling \({z}_{n}| {u}_{n},{s}_{n}={g}_{\theta }^{u\to z}({A}_{uz}{u}_{n}+{\gamma }_{uz},{s}_{n})\), in which Auz is a learned matrix of dimension Lz × L, and γuz is a bias vector of dimension Lz, where Lz is the dimension of zn. Without loss of generality, the remainder of the manuscript focuses on the case in which zn and un have the same dimension (L).
Modeling gene expression under technical effects
MrVI models the normalized expression of gene g, denoted hng, as a function of both zn and the nuisance covariate. This relationship is parameterized with multi-head attention (gθ above) to capture non-linear, nuisance-covariate-specific effects on gene expression. More information regarding this parameterization is available in ‘Additional model details’. Finally, we model the observed transcript counts with negative binomial distributions and account for the technical effects of the sequencing depth using the same approach as scVI22.
Variational approximation and training procedure
The generative model described by Equation (1) can be used to generate synthetic data; it does not directly inform on the posterior distribution of the latent variables un and zn, given observed gene expressions and sample ID of a given cell (n) required for analysis. Because we model zn as deterministic given un and sn, we must approximate the posterior pθ(un∣xn). Because this posterior term is intractable, we rely on variational inference to learn an approximation qϕ(un∣xn) to the posterior, in which ϕ denotes all parameters used to construct the variational approximation.
Modeling q ϕ(u n∣x n)
We model qϕ(un∣xn) as a Gaussian distribution whose mean and covariance (assumed diagonal) are outputs of multi-layer perceptrons (MLPs), fϕ, taking xn as inputs.
With this variational approximation, a straightforward approach to get posterior latent un and zn for a given cell consists of sampling un from qϕ(un∣xn), then computing zn as described in Equation (1).
Training procedure
We optimize the evidence lower bound (ELBO), which we maximize over the generative model parameters θ and variational parameters ϕ using mini-batch stochastic gradient descent methods56,57. In this problem, the ELBO writes as:
As mentioned previously, we also add a scaled L2 penalty to this objective on the distance between z and u. Thus, the final objective writes as
where c is a scalar coefficient set such that the penalty term matches the log probability of z for an isotropic Gaussian distribution centered around u.
Model architecture
Overall, MrVI uses a variety of MLPs, multi-head attention layers and learnable embeddings to estimate the parameters of the different variational and likelihood distributions described above (Extended Data Figure 8).
Exploratory analysis of sample effects on cell states
A common scenario in large-scale studies is that sample-level covariates are incomplete, noisy, or inconsistent across datasets. It may also be the case that the most relevant sample characteristics affecting gene expression are unobserved. Here, MrVI can identify the most relevant sources of heterogeneity between samples without assuming access to relevant target covariates (Fig. 1c); the most salient axes of variation across samples can then be related back to observed target covariates. This type of analysis relies on cell-state counterfactuals, which are used to quantify sample distances at the cellular level.
Predicting counterfactual cell states
After model fitting, MrVI can be used to predict the effect of a given sample on any cell. We aim to predict the counterfactual state of the cell, that is, its state had it been collected from another sample. We achieve this by substituting the sample-of-interest \({s}^{{\prime} }\ne {s}_{n}\) for the true sample-of-origin sn in Equation (1) to obtain
where un is the inferred cell state for cell n obtained through variational inference. \({z}_{n}^{{s}^{{\prime} }}\) captures the counterfactual state of cell n had it been collected from sample \({s}^{{\prime} }\).
Estimating sample distances at the cellular level
MrVI allows for unsupervised sample stratification by comparing distances between counterfactuals from Equation 2. In fact, we can assess the differences between samples sa and sb on a cell n by computing the distance between their respective counterfactual cell states \({z}_{n}^{{s}_{a}}\) and \({z}_{n}^{{s}_{b}}\). In particular, low distances between counterfactuals indicate that the two samples have similar effects on the cell according to the model.
On the basis of this observation, we summarize the sample stratification for a given cell as a sample distance matrix by computing the distance between counterfactuals for all pairs of samples. More precisely, for any cell n, we let D(n) denote its sample distance matrix between counterfactuals. In this matrix, the element at the position indexed by (sa, sb), where sa and sb are indices representing different samples, corresponds to the Euclidean distance between the counterfactual cell states \({z}_{n}^{{s}_{a}}\) and \({z}_{n}^{{s}_{b}}\).
These matrices inform sample stratification at single-cell resolution. They can first be used to identify cell populations with homogeneous sample stratifications. Clustering cells using their distance matrices as feature vectors can identify populations of cells with homogeneous sample stratifications. To do so, we embed each flattened distance matrix using PCA before clustering cells using the Leiden algorithm58. In any resulting cluster, we then assess sample stratification in aggregate. We first compute the average sample distance matrix of the cluster, which we then use to cluster samples using hierarchical clustering.
Owing to the uncertainty in un, even two samples with identical underlying distributions will have non-zero distances between their estimated counterfactual cell states. To account for this uncertainty, MrVI optionally computes Monte Carlo estimates of the distribution of distances between two counterfactual cell states derived from the same sample. These distribution estimates can then be used to determine the z score for the original distance matrix values. In detail, for a cell (xn), \({u}_{n}^{1},{u}_{n}^{2} \sim \hat{p}(u| {x}_{n})\) can be sampled, and the L2 distance between them can be computed, \(\parallel {u}_{n}^{1}-{u}_{n}^{2}{\parallel }_{2}\). The mean (\(\hat{\mu }\)) and the s.d. (\(\hat{\sigma }\)) of this term with more Monte Carlo samples can be estimated, and these can be used to compute normalized distances for D(n) as \(\frac{D(n)-\hat{\mu }}{\hat{\sigma }}\).
Assessing compositional and expressional sample differences
With observed sample covariates at hand, MrVI can also highlight which cells have different abundances or expression levels across groups (Fig. 1d). Such characteristics can, for instance, correspond to age, sex or disease status when samples correspond to different donors. The target covariate can also be derived from a stratification of samples based on the procedure described in the previous section. This section outlines how MrVI can be used for both DE and DA analyses assessing sample differences in gene expression and cell composition.
Cluster-free assessment of differences in expression
First, MrVI can characterize differential expression patterns across samples. Suppose we observe C target covariates in the form of a vector \({c}^{s}\in {{\mathbb{R}}}^{C}\) for each sample (s). To identify affected cells and genes by each target covariate, we fit the following linear model for each cell (n):
where \({z}_{n}^{1},\ldots {z}_{n}^{S}\) are S counterfactuals for cell n obtained from Equation (2). Here, \({\beta }_{n}\in {{\mathbb{R}}}^{C\times L}\) is the vector of regression coefficients obtained via least-squares regression.
Identifying the effect of covariates on cells
This linear model can first quantify the overall effect of an observed covariate on any cell. We compute, for any cell n and covariate index j ≤ C, the Chi-squared statistic of \({\beta }_{n}^{j}\). This statistic quantifies the extent to which the observed covariate j explains the variation in the counterfactual cell states.
Detecting cells strongly affected by covariate
The results of the linear regression can help identify cells strongly affected by a covariate. We compute the L2 norm of the vector βi for a covariate i. This yields effect strengths in the z representation for the specific covariates and can be used to compare effect strength across multiple cell types.
Detecting differentially expressed genes
The results of the linear regression can also identify DE genes associated with a given covariate in any cell. For simplicity, assume that the covariate of interest is binary. Let \({\beta }_{n}^{1}\in {{\mathbb{R}}}^{L}\) denote the regression coefficients of the covariate of interest for cell n. To identify the associated DE genes, we decode the counterfactual cell state \({z}_{n}^{1}={\beta }_{n}^{1}+{u}_{n}\) and the reference cell state \({z}_{n}^{0}={u}_{n}\). This computation yields two vectors of decoded gene expressions, denoted as \({h}_{n}^{1}\) and \({h}_{n}^{0}\). We then compute the log (fold change) between these two vectors, measuring the effect of covariate j on each of the observed genes g in the cell.
Accounting for out-of-distribution samples
Before conducting the described procedure, we first identify and discard samples that are out of distribution for any given cell. Samples will be out of distribution for a cell if no cell from that sample was collected in a similar cell state in the un space. For these samples, the model has insufficient information to accurately infer realistic counterfactual cell states in Equation (2). Thus, we conservatively discard the sample s for cell state u if the maximum density reached at u with respect to the approximate variational posterior distributions falls below a given threshold τ. More details on how the densities are computed and how τ is chosen are given in ‘Additional model details’.
Assessing cluster-free differences in composition
Last, our approach can identify differentially abundant cell populations over groups of samples using log ratios of aggregated posterior densities. For this purpose, we introduce qs, the aggregated posterior distribution for a given sample s, which corresponds to \({q}_{s}(u):= \frac{1}{{n}_{s}}{\sum }_{n:{s}_{n} = s}q_\phi(u| {x}_{n})\), where ns denotes the number of cells in the considered sample and q(u∣xn) is the variational approximation to the posterior distribution over the u space for cell n. We can then quantify the density of any set of samples A ⊂ {1, …S} in the u space as \({q}_{A}(u):= \frac{1}{| A| }{\sum }_{s\in A}{q}_{s}(u).\)
For two disjoint sets of samples A and B, we quantify the relative overabundance of cells from A compared with B at any cell state u by computing the log (density ratio) of the aggregated posterior densities of the two groups, that is:
We can then identify enriched or depleted regions of u in A compared with B by inspecting the log ratio rAB(u). This approach has several benefits. Given that MrVI assesses differential abundance in the u latent space, the captured differential-abundance effects are orthogonal to the differential-expression effects quantified in the previous section. Furthermore, this approach allows us to identify enriched cell states without cell-type or neighborhood assignments.
At the cluster level, we also devise a strategy to identify cell-enriched or depleted subpopulations with statistical confidence. For this purpose, letting A denote a cluster of interest, we collect log-ratios for (1) all cells in cluster A, and (2) those not in A. We then test for difference in the mean of these two sets of log ratios using a two-sided, two-sample t-test. To avoid detecting differences as significant owing to large sample sizes, the t-test rejects for the composite null that the mean difference is, in absolute value, less than a given threshold δ. Throughout the experiments, we set δ = 0.1.
Additional model details
Mixture of Gaussians prior for u n
MrVI posits a mixture of Gaussians (MoG) prior on un, that writes as:
Here, cn denotes the mixture component assigned to cell n. In practice, we assume the covariance matrices to be diagonal and learn μ1, …, μK, Σ1, …, ΣK, and π1, …, πK during training using maximum likelihood estimation.
When cell-type annotations are available, MrVI can weakly encourage the mixture of Gaussians to align with these annotations. In this case, we set K to be the number of unique cell-type annotations and reparameterize the mode of the Gaussian distribution as \({c}_{n} \sim {\rm{Categorical}}({\pi }_{1,n}^{{\prime} },{\pi }_{2,n}^{{\prime} },\ldots ,{\pi }_{K,n}^{{\prime} })\), in which \(\log {\pi }_{k,n}^{{\prime} }=\log {\pi }_{k}+\epsilon {\mathbb{I}}\left({y}_{n}=k\right)\). Here, ϵ is a positive constant (with a default of 10), yn is the cell-type annotation of cell n, and \({\mathbb{I}}(.)\) is the indicator function.
Parameterization of multi-head attention layers
Two components of MrVI rely on multi-head attention layers, corresponding to the mappings \({g}_{\theta }^{u\to z}\) and \({g}_{\theta }^{z\to h}\) from Equation (1). We now provide details on how these layers are parameterized, illustrated in Extended Data Figure 9.
Parameterization of \({g}_{\theta }^{u\to z}\)
\({g}_{\theta }^{u\to z}\) takes as inputs the cell state (un) and the sample from which the cell was collected (sn). We associate each sample ID s ∈ {1, …S} with an embedding \({e}_{s}\in {{\mathbb{R}}}^{L}\), learned during training. We then rely on a multi-head attention mechanism to capture the effect of sn on un in a non-linear fashion, considering un as queries and \({e}_{{s}_{n}}\) as keys or values. This output is then passed through a series of two fully connected layers, with ReLU activations, to obtain the actual output of \({g}_{\theta }^{u\to z}\).
Parameterization of \({g}_{\theta }^{z\to h}\)
\({g}_{\theta }^{z\to h}\) relies on the exact same parameterization as \({g}_{\theta }^{u\to z}\), but takes zn and bn as inputs instead of un and sn.
Out-of-distribution checks
The MrVI DE module actively filters for out-of-distribution cell–sample pairs. It might, for instance, be the case that a given sample contains no cells of a given type; in this case, this sample should be discarded for the DE analysis in Equation (3). We identify these out-of-distribution samples by defining the following admissibility score:
For a given cell n, we filter out sample s whenever As(u)≤τs, where τs is set to the 5% quantile of reference admissibility scores \({R}_{s}^{{\prime} }(n)\) over all of the cells collected in s, in which:
Notably, when computing the baseline admissibility scores over in-sample observations, we exclude the cell itself. After computing the set of admissible samples for every cell, additional filtering can be performed on the level of cells to eschew those with very few admissible samples (for example, a rare cell type observed in one sample), for which counterfactual estimates could be generally unreliable (see Supplementary Fig. 23).
Benchmark
Baselines
Exploratory analyses
We considered two approaches that stratify samples on the basis of differences in cell cluster abundance. Both approaches compare subcluster proportions between samples to yield distance matrices. More particularly, they subcluster each predefined cell group with the Leiden algorithm58 using low-dimensional cell representations, that is PCA for Composition (PCA) or scVI for Composition (SCVI). The scVI model was given the batch ID numbers to correct for batch effects in the latent representation. The PCA algorithm, however, does not have an explicit way to handle batch IDs. The distance between two arbitrary samples is then defined as the Euclidean distance between their subcluster proportions.
Guided analyses
We also considered Milo19 and miloDE61, which leverage estimates for DA and DE, respectively, in guided analyses. Milo is a statistical framework that aims to detect cell neighborhoods enriched in certain sample groups based on a nearest-neighbor graph of cells. Built on top of Milo, miloDE59 performs differential expression tests for each neighborhood identified by Milo by comparing each neighborhood against adjacent ones. These approaches, however, do not provide effect sizes for DA and DE at the cell level and instead group cells into neighborhoods that could obscure effect sizes at a single-cell resolution18. To compare these approaches to MrVI, we computed cell-level effect sizes for Milo and miloDE by defining cell-level effect sizes as the average effect size of the neighborhoods to which each cell belonged.
Metrics
Cell-type silhouette scores
We consider averaged silhouette width scores, computed as in ref. 30, to assess the relevance and the proper mixing of the latent representation u under the assumption that the same cell types appear across the considered samples. To do so, we first compute the silhouette score with respect to author-provided cell-type annotations. For any cell n with cell representation r(n), belonging to annotation Co, let d(n, C) denote the mean distance of r(n) to representations of annotation C, excluding n if C = Co. a(n) denotes the average distance of r(n) to cells of the same annotation, and b(n) the smallest mean distance of r(n). The silhouette score for cell n is computed as:
and the overall dataset silhouette score is the average of rescaled silhouette scores across all cells in the data. The rescaling, \(\tilde{s}(n)=\frac{1}{2}(s(n)+1)\), puts the dataset score in the range (0, 1). This score assesses to what extent the data representations cluster according to the annotations. When the dataset score is equal to 1, representations with the same annotation perfectly cluster together.
Batch silhouette scores
We also used the silhouette to measure the extent to which batch IDs mix together in the latent space. To do so, we follow the procedure described in ref. 30, which consists of, for each previously annotated cell type: (1) computing cell silhouette scores with respect to the batch assignments, (2) rescaling these scores, such that \(\hat{s}(n)=1-| s(n)|\), and (3) computing an overall silhouette score computed as a weighted average of \(\hat{s}(n)\), to ensure that each cell type gets the same contribution.
Data and preprocessing
We here provide a brief description of the datasets and of the preprocessing steps applied to them. More details can be found in Supplementary Note A.
Semi-synthetic experiment
We constructed a semi-synthetic dataset containing controlled DE and DA effects, on the basis of an original PBMC dataset of 68,000 cells29 (Extended Data Fig. 10). The synthetic dataset contains 32 synthetic study subjects, as well as five cell subsets, each of which can be seen as a cell type. In the first subset, denoted as A, there are differences in expression across subject groups characterized by covariate 1. Although we have no exact ground truth for the subject–subject distance in subset A, our ground truth consisted of a dendrogram, or tree, over study subjects, characterizing the similarities between subjects in terms of gene expression in the cell subset A. Specifically, all subjects sharing the same covariate 1 value shared similar gene expression values for cells in subset A. In two other cell subsets, denoted as B and C, cells had no differences in expression over subjects but exhibited differences in abundance, either corresponding to enrichment or depletion of these cell subsets in a specific group of samples. In particular, all subjects sharing the same covariate 2 value shared similar proportions of cell subsets B and C, but different proportions with the other subjects.
COVID-19 experiment
The original dataset7 contained a total of 650,000 PBMC cells sequenced across three sites: Cambridge, Sanger and Newcastle. We discarded cells coming from Cambridge and Sanger, and focused on data points sequenced in Newcastle. We retained the 10,000 most variable genes using Seurat v3. The resulting dataset contained 418,768 cells, originating from 55 individuals. No additional cell or gene filtering was performed.
sci-Plex experiment
The original sci-Plex dataset6 contains gene expression over three cell lines, exposed to 188 small-molecule drugs at four different doses. We filtered for drugs with no effects, resulting in a final dataset containing 251,088 cells with 92 drugs at all four doses and the vehicle cells. We applied highly variable gene selection using Seurat v3, retaining the top 5,000 genes.
Inflammatory bowel disease experiment
We downloaded the dataset42 from the Broad Institute Single Cell Portal, conducted in 46 individuals with the disease and 25 controls across tissue regions and sample preparation protocols, for a total of 463,000 cells. We retained the 10,000 most variable genes using Seurat v3.
Code Dependencies
MrVI was implemented using the scivi-tools framework with jax as the machine learning backend. The single-cell data was primarily stored as anndata structures, and xarray and pandas were used to store downstream model outputs. For preprocessing and visualization, we used scanpy, leidenalg, pymde, matplotlib, plotnine and seaborn. We used the scib-metrics package for computing various integration metrics.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data corresponding to the different experiments of this manuscript are publicly available. The semi-synthetic dataset, relying on ref. 29, can be downloaded at https://support.10xgenomics.com/single-cell-gene-expression/datasets/1.1.0/fresh_68k_pbmc_donor_a?. The COVID-19 dataset7 can be retrieved at https://covid19.cog.sanger.ac.uk/submissions/release1/haniffa21.processed.h5ad. The sciplex data6 is retrievable at https://figshare.com/ndownloader/files/33979517. The IBD dataset can be downloaded at https://singlecell.broadinstitute.org/single_cell/study/SCP1884/human-cd-atlas-study-between-colon-and-terminal-ileum.
Code availability
MrVI is available within scvi-tools (https://scvi-tools.org/). The version of MrVI used to run the data analyses is deposited in a standalone, now deprecated GitHub package: https://github.com/YosefLab/mrvi/tree/paper_reproducibility. All code and dependencies required to reproduce the data analysis can be found at https://github.com/YosefLab/mrvi-reproducibility/tree/v0.1.0. The code used to produce the figures in this manuscript has been deposited via Zenodo at https://doi.org/10.5281/zenodo.14927976 (ref. 60).
References
Sanchez-Vega, F. et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 173, 321–337 (2018).
GTEx Consortium. Genetic effects on gene expression across human tissues. Nature 369, 1318–1330 (2017).
Kang, H. M. et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 36, 89–94 (2018).
McGinnis, C. S. et al. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat. Methods 16, 619–626 (2019).
Smillie, C. S. et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 178, 714–730 (2019).
Srivatsan, S. R. et al. Massively multiplex chemical transcriptomics at single-cell resolution. Science 367, 45–41 (2020).
Stephenson, E. et al. Single-cell multi-omics analysis of the immune response in COVID-19. Nat. Methods 27, 904–916 (2021).
Tran, V. et al. High sensitivity single cell RNA sequencing with split pool barcoding. Preprint at bioRxiv https://doi.org/10.1101/2022.08.27.505512 (2022).
Yazar, S. et al. Single-cell eQTL mapping identifies cell type-specific genetic control of autoimmune disease. Science 376, eabf3041 (2022).
Velmeshev, D. et al. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689 (2019).
Perez, R. K. et al. Single-cell RNA-seq reveals cell type-specific molecular and genetic associations to lupus. Science 376, eabf1970 (2022).
Hoadley, K. A. et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304 (2018).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Heumos, L. et al. Best practices for single-cell analysis across modalities. Nat. Rev. Genet. 24, 550–572 (2023).
Eraslan, G. et al. Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Science 376, eabl4290 (2022).
Salcher, S. et al. High-resolution single-cell atlas reveals diversity and plasticity of tissue-resident neutrophils in non-small cell lung cancer. Cancer Cell 40, 1503–1520 (2022).
Mitchel, J. et al. Coordinated, multicellular patterns of transcriptional variation that stratify patient cohorts are revealed by tensor decomposition. Nat. Biotechnol. 43, 1192–1201 (2025).
Maity, A. K. & Teschendorff, A. E. Cell-attribute aware community detection improves differential abundance testing from single-cell RNA-seq data. Nat. Commun. 14, 3244 (2023).
Dann, E. et al. Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol. 40, 245–253 (2022).
Abdollahi, A. et al. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J. Exp. Med. 201, 925–935 (2005).
Burkhardt, D. B. et al. Quantifying the effect of experimental perturbations at single-cell resolution. Nat. Biotechnol. 39, 619–629 (2021).
Lopez, R. et al. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Boyeau, P. et al. An empirical Bayes method for differential expression analysis of single cells with deep generative models. Proc. Natl Acad. Sci. USA 120, e2209124120 (2023).
Lotfollahi, M., Wolf, F. A. & Theis, F. J. scGen predicts single-cell perturbation responses. Nat. Methods 16, 715–721 (2019).
Lotfollahi, M. et al. Predicting cellular responses to complex perturbations in high-throughput screens. Mol. Syst. Biol. 19, e11517 (2023).
Weinberger, E., Lin, C. & Lee, S.-I. Isolating salient variations of interest in single-cell data with contrastiveVI. Nat. Methods 20, 1336–1345 (2023).
Gayoso, A. et al. A Python library for probabilistic analysis of single-cell omics data. Nat. Biotechnol. 40, 163–166 (2022).
Kingma, D. P. & Welling, M. Auto-encoding variational bayes. Preprint at arXiv https://doi.org/10.48550/arXiv.1312.6114 (2013).
Zheng, G. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Luecken, M. D. et al. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods 19, 41–50 (2022).
Schulte-Schrepping, J. et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell 182, 1419–1440 (2020).
Martinez, F. O., Combes, T. W., Orsenigo, F. & Gordon, S. Monocyte activation in systemic Covid-19 infection: assay and rationale. EBioMedicine 59, 102964 (2020).
Shojaei, M. et al. IFI27 transcription is an early predictor for COVID-19 outcomes, a multi-cohort observational study. Front. Immunol. 13, 1060438 (2023).
de Cevins, C. et al. A monocyte/dendritic cell molecular signature of SARS-CoV-2-related multisystem inflammatory syndrome in children with severe myocarditis. Med 2, 1072–1092 (2021).
Subramanian, A. et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171, 1437–1452 (2017).
Jost, M. et al. Pharmaceutical-grade rigosertib is a microtubule-destabilizing agent. Mol. Cell 79, 191–198 (2020).
Bagratuni, Tina et al. JQ1 inhibits tumour growth in combination with cisplatin and suppresses JAK/STAT signalling pathway in ovarian cancer. Eur. J. Cancer 126, 125–135 (2020).
Shi, J. & Walker, M. G. Gene set enrichment analysis (GSEA) for interpreting gene expression profiles. Curr. Bioinform. 2, 133–137 (2007).
Liberzon, A. et al. The molecular signatures database hallmark gene set collection. Cell Syst. 23, 417–425 (2015).
Horwitz, K. B., Costlow, M. E. & McGuire, W. L. MCF-7: a human breast cancer cell line with estrogen, androgen, progesterone, and glucocorticoid receptors. Steroids 26, 785–795 (1975).
Grosveld, G. et al. The chronic myelocytic cell line K562 contains a breakpoint in bcr and produces a chimeric bcr/c-abl transcript. Mol. Cell. Biol. 6, 607–616 (1986).
Kong, L. et al. The landscape of immune dysregulation in Crohn’s disease revealed through single-cell transcriptomic profiling in the ileum and colon. Immunity 56, 444–458 (2023).
Hashash, J. G. et al. Altered expression of the epithelial mucin MUC1 accompanies endoscopic recurrence of post-operative Crohn’s disease. J. Clin. Gastroenterol. 55, 127–133 (2021).
Leal, R. F. et al. Identification of inflammatory mediators in patients with Crohn’s disease unresponsive to anti-TNFα therapy. Gut 64, 233–242 (2014).
Pavlidis, P. et al. Interleukin-22 regulates neutrophil recruitment in ulcerative colitis and is associated with resistance to ustekinumab therapy. Nature Commun. 13, 5820 (2022).
Romano, E., Rosa, I., Fioretto, B. S. & Manetti, M. The contribution of endothelial cells to tissue fibrosis. Curr. Opin. Rheumatol. 36, 52–60 (2024).
Mukherjee, P. K. et al. Stricturing Crohn’s disease single-cell RNA sequencing reveals fibroblast heterogeneity and intercellular interactions. Gastroenterology 165, 1180–1196 (2023).
Pilling, D., Vakil, V., Cox, N. & Gomer, R. H. TNF-α-stimulated fibroblasts secrete lumican to promote fibrocyte differentiation. Proc. Natl Acad. Sci. USA 112, 11929–11934 (2015).
Yang, K. et al. Transforming growth factor-β induced protein regulates pulmonary fibrosis via the G-protein signaling modulator 2/Snail axis. Peptides 155, 170842 (2022).
Wu, F. & Chakravarti, S. Differential expression of inflammatory and fibrogenic genes and their regulation by NF-κB inhibition in a mouse model of chronic colitis. J. Immunol. 179, 6988–7000 (2007).
Rieder, F. et al. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J. Pathol. 179, 2660–2673 (2011).
Siletti, K. et al. Transcriptomic diversity of cell types across the adult human brain. Science 382, eadd7046 (2023).
Kumar, D., Cinghu, S., Oldfield, A. J., Yang, P. & Jothi, R. Decoding the function of bivalent chromatin in development and cancer. Genome Res. 31, 2170–2184 (2021).
Takahashi, H., Iwata, T., Yamanaka, Y., Yamada, M. & Yagi, S. Variational autoencoder with implicit optimal priors. In Proc. AAAI Conference on Artificial Intelligence 5066–5073 (Association for the Advancement of Artificial Intelligence, 2019).
Ding, J. & Regev, A. Deep generative model embedding of single-cell RNA-seq profiles on hyperspheres and hyperbolic spaces. Nat. Commun. 12, 2554 (2021).
Kingma, D. P. & Ba, J. Adam: A method for stochastic optimization. Preprint at arXiv https://doi.org/10.48550/arXiv.1412.6980 (2014).
Yao, Y., Rosasco, L. & Caponnetto, A. On early stopping in gradient descent learning. Constr. Approx. 26, 289–315 (2007).
Traag, V. A., Waltman, L. & van Eck, N. J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 5233 (2019).
Missarova, A., Rosen, L. U., Dann, E., Satija, R. & Marioni, J. Leveraging neighborhood representations of single-cell data to achieve sensitive DE testing with miloDE. Genome Biol. 5, 189 (2024).
Boyeau, P., Hong, J. & Ergen, C. MrVI manuscript reproducibility code. Zenodo https://doi.org/10.5281/zenodo.14927976 (2025).
Acknowledgements
We thank L. Shi, F. Ingelfinger, F. Sheban, N. Levy, R. Giglio, N. Hou, K. Hoffer-Hawlik, S. Kevlishvili and A. Steinberg for being the first to try the MrVI Python package and providing valuable feedback that greatly improved our work. This work was supported by a Chan-Zuckerberg Initiative Seed Networks for the Human Cell Atlas grant (CZF2019-002452) and NIAID Grant R01 AI169075 and R24 AI145816 to N.Y. Grants 2022-253560 (for J.H.), 2021-237773 and 2024-342826 from the Chan Zuckerberg Initiative DAF, an advised fund of the Silicon Valley Community Foundation. C.E. was supported by a Walter-Benjamin postdoctoral fellowship by the DFG (448802458).
Author information
Authors and Affiliations
Contributions
P.B. and J.H. contributed equally to this work. P.B., J.H., A.G. and N.Y. conceived the original research questions and developed the conceptual framework. P.B., J.H., A.G. and C.E. designed the methodology. P.B., J.H., A.G. and C.E. developed the software and conducted experimental analyses. M.K. and J.H. implemented the reproducibility pipeline and re-engineered MrVI for integration with the scvi-tools package. P.B., J.H. and C.E. performed the primary experimental analyses presented in the manuscript. J.L.M. and E.A. provided domain expertise for the sci-Plex dataset analysis. C.E. and N.Y. jointly supervised the project and guided the overall research direction. M.I.J. and E.A. provided additional supervisory support for specific aspects of the work. P.B., J.H., A.G., M.K., J.L.M., M.I.J., E.A., C.E. and N.Y. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
A.G. is currently an employee of Google DeepMind. Google DeepMind has not directed any aspect of this study nor exerts any commercial rights over the results. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks Qi Liu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer review information: Primary Handling Editor: Lin Tang, in collaboration with the Nature Methods team.Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Comparison oftime since symptom onset for the two COVID-19 patient clusters from Figure 3d.
These differences are significant under a Mann-Whitney U test (p < 0.05). We find several outliers, which might be attributed to the exact course of disease, or to noise due to the fact that symptom onset is self-reported. This ambiguity can’t be resolved with the current data. In this and all other plots where box plots are used, they indicate the median (center lines), interquantile range (hinges), and 5th to 95th percentiles (whiskers).
Extended Data Fig. 2 Sample distances of all 369 samples used in the analysis for the A549 cell line.
The columns are annotated by each drug’s pathway annotation from the original study, dosage level, and whether the sample was retained for the remaining analysis (top 20 percent of samples based on distance from vehicle). The hierarchical clustering was performed with the Ward variance minimization algorithm.
Extended Data Fig. 3 Analysis of the MCF-7 cell line in the sci-Plex experiment.
MrVI was fit over 92 drugs each at four doses that passed the DE-gene filter. The analysis is performed in a way similar to Figure 4. a. and b. MDEs of the u and z latent spaces colored by the pathway of the drug used to treat each cell (left) and the cell cycle stage of each cell (right). For the MDEs colored by pathway, only the top 20 percent of samples based on distance from the vehicle are shown in full opacity. c. PCA of sample distance matrices. Left: scatterplot of all local sample distance matrices projected onto the top two principal components colored by cell-cycle stage displays no visual subclusters. Right: barplot of the proportion of variance explained against the number of principal components used. d. Comparison of MrVI against the benchmark methods based on performance metrics assessing alignment with prior knowledge. Each bar represents the metric for one model fit, except for ‘Random’ which reports the 95% confidence interval over 100 permutations of the inferred distance matrix from MrVI. Left: average percentile of intra-drug distances, measuring how much closer samples with the same drug and different doses are to each other relative to other samples (lower is better). Right: silhouette score of sample clusters with similarities inferred from DEG sets in the Connectivity Map dataset, assessing cluster consistency (higher is better). e. Hierarchically clustered sample distance matrix. Rows are annotated by the pathway, dose, and cluster of each sample (clusters inferred from the distance matrix). For e. and f., the analysis is performed over the top 20 percent of drug-dose combinations (74/368) based on their distance from the vehicle. f. Heatmap of Gene Set Enrichment Analysis (GSEA) scores for the Human MSigDB Hallmark gene set collection for DE genes identified for each cluster in panel e. Each tile’s upper-right and bottom-left triangles, respectively, represent scores for the set of up-regulated and down-regulated DE genes. MCF-7 is a hormone-receptor-positive breast cancer cell line and was the only cell line to exhibit strong effects in response to the hormone therapies, fulvestrant and toremifene citrate, as evident in both the z space and the sample distance matrix.
Extended Data Fig. 4 Analysis of the K562 cell line in the sci-Plex experiment.
MrVI was fit over 92 drugs each at four doses that passed the DE-gene filter. The analysis is performed in a way similar to Figure 4. a. and b. MDEs of the u and z latent spaces colored by the pathway of the drug used to treat each cell (left) and the cell cycle stage of each cell (right). For the MDEs colored by pathway, only the top 20 percent of samples based on distance from the vehicle are shown in full opacity. c. PCA of sample distance matrices. Left: scatterplot of all local sample distance matrices projected onto the top two principal components colored by cell-cycle stage displays no visual subclusters. Right: barplot of the proportion of variance explained against the number of principal components used. d. Barplot comparing MrVI against the benchmark methods for a performance metric that determines alignment with prior knowledge. Each bar represents the metric for one model fit, except for ‘Random’ which reports the 95% confidence interval over 100 permutations of the inferred distance matrix from MrVI. The average percentile of distances measures how much closer samples with the same drug and different doses are to each other relative to the rest of the distances. We expect the average percentile to be low. There was no available Connectivity Map data for the K562 cell line, so we could not compute the silhouette metric for this dataset. e. Hierarchically clustered sample distance matrix. Rows are annotated by the pathway, dose, and cluster of each sample (clusters inferred from the distance matrix). For e. and f., the analysis is performed over the top 20 percent of drug-dose combinations (74/368) based on their distance from the vehicle. f. Heatmap of Gene Set Enrichment Analysis (GSEA) scores for the Human MSigDB Hallmark gene set collection for DE genes identified for each cluster in panel e. Each tile’s upper-right and bottom-left triangles, respectively, represent scores for the set of upregulated and down-regulated DE genes. We observe a cluster of Bcr-Abl tyrosine kinase inhibitors (bosutinib, dasatinib, nilotinib) with a significant effect, which is absent from the other cell lines.
Extended Data Fig. 5 SCIB metrics computed on the IBD dataset.
Tissue (colon or ileum) was used as batch key, and the original study’s annotations were used as cell-type labels as displayed in Extended Data Figure 6. MrVI (CT prior) uses the cell-type specific bias for the mixture weights using the existing annotations (Methods), while MrVI is the default version relying on mixture of Gaussians without cell-type biases.
Extended Data Fig. 6 Additional analysis of stromal cells in stenosis.
a. UMAP colored by all cell types in the original study as well as colored by disease behavior. b-e UMAP subset to stromal cells. b. colored by disease behavior. c. Same display as in Figure 5a of effect size of disease behavior B2 highlighting high score in a subset of pericytes. d. Raw gene expression after library-size normalization and log1p-transformation for marker genes of endothelial-to-mesenchymal transition. e. Additional predicted LFCs in patients with stenosing course of disease (B2) versus patients with B1 disease behavior patients estimated by MrVI.
Extended Data Fig. 7 Additional analysis of endothelial cells in stenosis.
a. Violin plots illustrating changes in normalized gene expression between inflamed and non-inflamed biopsies from patients categorized as B1, B2, and B3 disease behavior after subsetting to Pericytes HIGD1B STEAP4. Raw gene expression values are normalized by library size and log1p-transformed. b. Dotplots displaying top 3 marker genes for the different endothelial cell (EC) subclusters. Lymphatic endothelial cells were excluded from this analysis. c. Differential abundance from Figure 5c displayed as violin plots to demonstrate changes in abundance across cell types. Displayed is the log density ratio between samples from B2 and B1 disease behavior. We report the significance of the hypothesis that the difference of log density ratios between a given cell type and all other cell types is above 1 in absolute value (see Methods). The number of cells of each type is reported in Supplementary Table 4.
Extended Data Fig. 8 Functional description of MrVI.
The encoder, which approximates the posterior distribution, is modeled with a multi-layer perceptron, while the decoder, which approximates the generative process, is composed of two attention blocks: one which incorporates the sample identifiers and one which incorporates the batch identifiers.
Extended Data Fig. 9 Illustration of the parameterization of the multi-head attention mechanism used in MrVI.
For any cell n, this mechanism takes an embedding and a latent representation as inputs. These two inputs respectively serve as keys/values and queries for the attention mechanism. This output is then passed through a series of fully connected layers to obtain the final output.
Extended Data Fig. 10 Cartoon of semi-synthetic benchmark construction presented in Figure 2.
Data from a PBMC dataset of 68K cells [29] are clustered using the Leiden algorithm. Then, for cluster A, synthetic DE effects are introduced by assigning subclusters of cells to different study subjects. For clusters B and C, different proportions of each cell type are sampled depending on the study subject, producing a ground truth DA effect.
Supplementary information
Supplementary Information (download PDF )
Supplementary Notes A and B, Supplementary Figures 1–25 and Supplementary Tables 1–4.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Boyeau, P., Hong, J., Gayoso, A. et al. Deep generative modeling of sample-level heterogeneity in single-cell genomics. Nat Methods 22, 2264–2274 (2025). https://doi.org/10.1038/s41592-025-02808-x
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41592-025-02808-x
This article is cited by
-
Toward informed batch correction for single-cell transcriptome integration
Nature Computational Science (2026)
