
Abstract
During development, cells differentiate through a hierarchy of increasingly restricted cell types, a process that is summarized by a cell differentiation map. Recent technologies profile lineages and cell types at scale, but existing methods to infer cell differentiation maps from these data rely on heuristic models with restrictive assumptions about the developmental process. Here we introduce a quantitative framework to evaluate cell differentiation maps and develop an algorithm, called Carta, that infers an optimal differentiation map from single-cell lineage tracing data. The key insight in Carta is to balance the tradeoff between the complexity of the map and the number of unobserved cell type transitions on the lineage tree. We show that, in models of mammalian trunk development and mouse hematopoiesis, Carta identifies important features of development that are not revealed by other methods, including convergent differentiation of cell types, progenitor differentiation dynamics and new intermediate progenitors.
Similar content being viewed by others


Main
Organismal development occurs via the differentiation of cells through a hierarchy of ‘progenitor cell types’, each with progressively restricted potential, ultimately leading to specialized cell types. The ‘cell differentiation map’ describes this hierarchy, including all progenitor and specialized cell types and the transitions between these cell types. Deriving cell differentiation maps—of tissues, organs or complete organisms—is a key challenge in developmental biology.
The traditional method to derive cell differentiation maps involves manual lineage tracing that directly tracks cell division and differentiation during development1,2,3,4,5. A notable milestone using this approach was the derivation of the complete differentiation map of the 671 cells of Caenorhabditis elegans using timelapse microscopy6. However, such a direct observational approach is not feasible for more complex organisms, such as mice or humans, which contain trillions of cells and develop in utero.
More recently, single-cell RNA sequencing (scRNA-seq), which measures the transcriptomes of individual cells, has allowed investigation of cell differentiation maps at scale7,8,9,10,11,12. Cell differentiation maps are derived from these data using trajectory inference methods that attempt to infer branching structures and pseudotimes underlying dynamic differentiation processes from transcriptomes measured at one or a small number of timepoints13,14,15,16,17,18,19,20,21,22,23. These methods rely on several limiting assumptions that hinder their ability to reconstruct precise cellular relationships, particularly the assumption that all progenitor cell types along the differentiation hierarchy are observed in the data24,25.
Recent advances in genome editing and single-cell sequencing have enabled high-throughput lineage tracing of cells in complex developmental systems26,27,28,29. In these technologies, heritable barcodes are induced in dividing cells using genome editing tools such as CRISPR–Cas9, providing markers of cell divisions. The barcodes can be introduced either at specific stages of development30,31,32,33 or dynamically through a continuous process as cells divide and differentiate34,35,36,37,38,39. scRNA-seq simultaneously measures barcodes (revealing the lineage of cells) and gene expression (revealing cell types) for thousands of individual cells as the system develops40,41,42,43,44,45,46. These barcoding systems offer the scalability to investigate development in complex organisms but have limited resolution compared to exhaustive microscopy methods, such as those used for C. elegans. Thus, with these technologies, one does not typically observe the differentiation decisions of each dividing cell during development.
Current approaches of cell differentiation inference are based on two opposing assumptions about the progenitor cell types that exist in the developmental system. First, trajectory inference-based methods that infer cell differentiation maps from scRNA-seq data assume that all progenitor cell types are observed24,25. On the opposite extreme, other recent studies28,39,47 use distance-based heuristics calculated from single-cell lineage tracing data that implicitly assume that the cell differentiation map is a binary tree, and, consequently, the number of progenitor cell types is exactly one less than the number of observed cell types. Neither of these assumptions is likely to be true in practice; for example, early transient progenitor cell types that arise long before cell collection are likely unobserved, and the cell differentiation map is not always a tree due to phenomena such as alternate routes of differentiation to cell types (convergent differentiation)28. Given that current approaches of cell differentiation map inference vary greatly in their heuristics and assumptions, there is a need for a formal framework that can systematically assess varying models of cellular differentiation.
Here we provide a formal definition of an optimal cell differentiation map and derive an algorithm, called Carta, that infers an optimal cell differentiation map from single-cell lineage tracing data. We represent a cell differentiation map by a directed acyclic graph (DAG) whose vertices are cell types and whose edges represent transitions (differentiation events) between cell types that occur during development. Notably, our framework does not assume that all progenitor cell types are measured at the time of the experiment. Instead, we introduce the concept of a ‘potency’ set, defining unobserved progenitors by the cell types of their descendants. Using the concept of potency, we demonstrate that there are two competing objectives when inferring a cell differentiation map from lineage tracing data: the ‘complexity’ of the cell differentiation map and the ‘discrepancy’ between transitions in the map and the cell lineage tree. In computing the optimal cell differentiation map and discrepancy for any number of progenitor cell types, Carta quantifies the tradeoff between these objectives and provides a quantitative framework to evaluate different models of cell differentiation map inference.
The cell differentiation map inferred by Carta is both interpretable and recapitulates established developmental trajectories. On simulated cell differentiation maps and lineage tracing data, Carta more accurately reconstructs the underlying cell differentiation maps compared to existing methods. In an in vitro model for mammalian trunk development29,48,49, Carta infers a cell differentiation map that provides insights into the differentiation dynamics of neuromesodermal progenitors (NMPs) into somitic and neural tube lineages that are not revealed under the restricted frameworks of existing methods. On lineage tracing data from a mouse hematopoiesis model30, Carta infers a cell differentiation map that better recapitulates the established differentiation of hematopoiesis and also has stronger agreement with gene expression compared to existing methods. The framework of cell differentiation map inference presented here extends beyond the restrictions of existing methods and provides opportunities to better understand development in a variety of contexts.
Results
Carta: a framework for cell differentiation mapping
Carta infers an optimal cell differentiation map from one or more cell lineage trees while accounting for ambiguities arising from incomplete sampling and other limitations of current lineage tracing technologies (Fig. 1a,b). The inputs to Carta are ‘mcell lineage trees’ \({\mathcal{T}}:=\{{T}_{1},\ldots ,{T}_{m}\}\), with each tree Ti describing the cell division history of a distinct biological replicate of the same developmental system. The leaves of each tree correspond to sequenced cells labeled by their cell type, typically derived from scRNA-seq data, and internal vertices represent ancestral cells that are unlabeled, because the cell type of these cells is not measured (Fig. 1a). Let S be the set of ‘observed cell types’—that is, the set of cell types that label the leaves of \({\mathcal{T}}\).

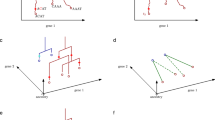
a, The input to Carta is one or more cell lineage trees, whose leaves are labeled by the measured cell type (labeled A, B and C) of the sequenced cells. Typically, some cells that are present at the time of the experiment are not sampled (denoted by dotted lines). b, Carta infers a cell differentiation map that describes the progenitor cell types—represented as a potency set—and cell type transitions that occurred during development. c, Carta quantifies the tradeoff between the number k of progenitor cell types in the cell differentiation map and its discrepancy with the cell lineage trees by computing the Pareto front of optimal solutions. A cell differentiation map with the optimal number k* of progenitors is chosen by identifying an elbow of the Pareto front.
A cell differentiation map F is a directed graph where the vertices represent cell types and the edges describe the transitions between cell types that occurred during development (Fig. 1b). These transitions are directly revealed by cell lineage trees that have all vertices labeled by cell type. However, typically, the cell types of ancestral cells are unknown. Moreover, the cell types of ancestral cells may not be observed on the leaves of the trees in \({\mathcal{T}}\).
The first key insight in Carta is that unobserved progenitor cell types are described by the set of observed cell types that the progenitor can differentiate into—that is, the possible future cell types of the descendants of that progenitor. We deem this set the potency of that progenitor. Formally, if S is the set of observed cell types, then the potency of a progenitor cell type is a subset of S. For instance, the totipotent cell—a progenitor that can differentiate into any observed cell type—has potency S, whereas an observed cell type t has potency {t}. We define a cell differentiation map FS for the set S of observed cell types to be a directed graph whose vertices represent observed cell types and unobserved progenitors—and are labeled by either an element of S or a subset of S—and whose edges represent cell type transitions that occurred during development (Fig. 1b).
The second key insight in Carta is a discrepancy score \(D({\mathcal{T}},{F}_{S})\) to evaluate the different cell differentiation maps FS that explain the development of observed cell types from a collection \({\mathcal{T}}\) of cell lineage trees. The discrepancy score \(D({\mathcal{T}},{F}_{S})\) quantifies the ‘fit’ between a candidate cell differentiation map FS and the observed cell types S on the leaves of cell lineage trees \({\mathcal{T}}\) (Methods and Supplementary Section 1). The discrepancy score measures how well the potency of a cell in the cell lineage tree (from a labeling induced by the candidate cell differentiation map) matches the ‘observed potency’—that is, the observed cell types of the cell’s descendants. A lower discrepancy score indicates a better fit between the cell differentiation map and the cell lineage trees \({\mathcal{T}}\) under the assumption that the cells in \({\mathcal{T}}\) follow the routes of differentiation in the map.
Carta infers a cell differentiation map FS from cell lineage trees \({\mathcal{T}}\) that balances the tradeoff between the complexity of FS, measured as the number k of progenitor cell types in FS, and its discrepancy score \(D({\mathcal{T}},{F}_{S})\). The least complex cell differentiation map (k = 1) has a single totipotent progenitor cell type that can differentiate into any observed cell type. However, this map will typically have a very high discrepancy score (Fig. 1c, upper left). On the other extreme, one can always find a cell differentiation map with minimum discrepancy \(D({\mathcal{T}},{F}_{S})=0\), but this map will often have a large number k of progenitor cell types, several of which may be false positives (Fig. 1c, bottom right). Carta solves the Cell Differentiation Map Inference Problem (CDMIP) (Methods), deriving a cell differentiation map FS with minimum discrepancy \(D({\mathcal{T}},{F}_{S})\) for each number k of progenitor cell types. These solutions give the ‘Pareto front’ of optimal solutions—that is, there are no cell differentiation maps that have both fewer number of progenitors and lower discrepancy compared to these solutions. Thus, Carta provides a systematic framework to evaluate cell differentiation maps with varying number of progenitors and to identify an optimal cell differentiation map with k* progenitors (Methods).
Simulated data
We compare Carta to two existing methods to infer cell differentiation maps from lineage tracing data, ICE-FASE47 and evolutionary coupling (EvoC)39, on simulated data. ICE-FASE and EvoC use distance-based heuristics calculated from cell lineage trees to perform hierarchical clustering of the cell types to produce the cell differentiation map. Although Carta relies only on the topology of the input cell lineage trees, both ICE-FASE and EvoC additionally require timed cell lineage trees as input. We apply two modes of Carta: Carta-Tree, in which the inferred cell differentiation map is a tree (not necessarily binary), and Carta-DAG, in which the inferred cell differentiation map is a DAG (Methods).
We simulated lineage trees under three types of ground truth cell differentiation maps: (1) binary trees with ∣S∣ = 8, 12, 16; (2) trees that have polytomies—that is, nodes with more than two children, with ∣S∣ = 8, 12, 16; and (3) DAGs, with ∣S∣ = 6, 10, 14 (Methods). For each cell differentiation map, we generated leaf-labeled cell lineage trees using a generalized birth–death model50 for cell divisions and a Markov process for cell type transitions. We sampled 50, 100 or 200 cells of each observed cell type uniformly at random from a larger tree to mimic sampling limitations of current technologies where only a fraction (approximately 10%) of the cells are sampled for sequencing (Methods)29. We generated five instances for each set of simulation parameters. We also included simulated trees provided in the ICE-FASE study (Fang et al.)47, although the lack of asymmetric division51,52 in their simulations led to trees with uninformative observed potencies (Supplementary Section 7 and Supplementary Fig. 1). We evaluated the performance of each method by comparing the ground truth set \({{\mathcal{P}}}^{* }\) of progenitors and the set \({\mathcal{P}}\) of inferred progenitors using two metrics: the Jaccard distance \({d}_{J}({\mathcal{P}},{{\mathcal{P}}}^{* })\) (ref. 53) between \({{\mathcal{P}}}^{* }\) and \({\mathcal{P}}\) and the normalized minimum Hamming distance, \({d}_{H}({\mathcal{P}},{{\mathcal{P}}}^{* })\), which evaluates the deviation of each progenitor in \({{\mathcal{P}}}^{* }\) from each progenitor in \({\mathcal{P}}\) (Methods). We additionally examine the precision and recall of the progenitors inferred by each method.
Carta consistently outperforms ICE-FASE and EvoC across most simulation parameters, with similar performance to ICE-FASE only when the cell differentiation map is a binary tree. Specifically, for the binary tree case (Fig. 2a), both Carta-Tree and ICE-FASE yield almost perfect reconstruction of the differentiation maps (median Jaccard distance 0), whereas Carta-DAG performs worse (median 0.231) but still outperforms EvoC (0.308). When the cell differentiation map is a tree with polytomies (Fig. 2b), Carta-Tree achieves the best results (median Jaccard 0.222), outperforming Carta-DAG (0.333), ICE-FASE (0.333) and EvoC (0.462). This demonstrates the strength of Carta-Tree in reconstructing general tree-structured differentiation maps beyond the restrictive binary tree assumption made by existing methods (ICE-FASE and EvoC). For DAG-structured differentiation maps (Fig. 2c), Carta-DAG (median Jaccard 0.332) outperforms Carta-Tree (0.455), ICE-FASE (0.5) and EvoC (0.615). Similar trends are consistent across other evaluation metrics (Supplementary Figs. 2–4). We additionally compared the performance of Carta and ICE-FASE on the simulated cell lineage trees presented in that study (Supplementary Section 7 and Supplementary Fig. 5). Using Carta’s heuristic mode, the largest instances (200 cells per cell type and 14 cell types) are completed with a mean of 736.35 seconds (Supplementary Section 8 and Supplementary Fig. 6), with the runtime being independent of the size of the differentiation maps (Supplementary Fig. 7).

a–c, Jaccard distance between the set of progenitors inferred by Carta-Tree, Carta-DAG, ICE-FASE and EvoC and ground truth when the cell differentiation map is a binary tree (a), a tree with polytomies (b) and a DAG (c). Each point represents the results for one of the 900 simulated cell lineage trees for each type of cell differentiation map (2,700 total). Results are shown for tree-structured cell differentiation maps with 12 observed cell states and DAG-structured cell differentiation maps with 10 observed cell states. Box plots show the median (in red) and the interquartile range (IQR), and the whiskers denote the lowest and highest values within 1.5 times the IQR from the first and third quartiles, respectively. d, Normalized discrepancy \(\tilde{D}({\mathcal{T}},{F}_{S})\) of cell differentiation maps, inferred by Carta-Tree (blue) and Carta-DAG (orange), for k = 1, … 8 progenitors reveals the Pareto fronts—here shown for a simulated instance with six progenitor cell types, six observed cell types and 200 cells of each observed cell type. Discrepancy of cell differentiation maps is inferred by ICE-FASE (purple) and EvoC (green) for their fixed number of five progenitors.
Carta identified the Pareto-optimal differentiation maps with different number k of progenitors (Fig. 2d). Carta-Tree guarantees lower or equal discrepancy of the inferred cell differentiation maps compared to ICE-FASE and EvoC, whereas Carta-DAG guarantees even lower discrepancy of the inferred maps by relaxing the tree constraints. Whereas ICE-FASE and EvoC are restricted to inferring maps with ∣S∣ − 1 progenitors, Carta supports a broad range of number of progenitors. Note that here and for the rest of the paper, we report the ‘normalized discrepancy’ \(\tilde{D}({\mathcal{T}},{F}_{S})\), which is the discrepancy divided by the total number of ancestral cells across the input lineage trees.
Cell differentiation mapping of trunk-like structures
We compared Carta and several other methods in inferring the routes of differentiation during mammalian trunk development. Specifically, we applied Carta (runtime: Supplementary Table 1), Fitch, PhyloVelo54, ICE-FASE47 and EvoC38 (Methods) to cell lineage trees derived from single-cell CRISPR–Cas9-based lineage tracing of an in vitro embryoid model called trunk-like structures (TLSs)29. TLSs mirror post-occipital mammalian trunk development and are particularly suited for studying the differentiation dynamics of NMP cells. NMPs are a pool of self-renewing progenitors that differentiate into both the neural tube, which forms the future spinal cord, and the flanking somitic mesoderm, which forms future vertebrae and muscle cells of the trunk (Fig. 3a)48. Given their bipotent nature, NMPs are particularly interesting as they produce cells of two germ layers in the posterior embryo, the neuroectoderm and the paraxial mesoderm, that are classically considered to come from separate origins55,56,57. This dataset consists of 14 cell lineage trees with a total of 6,570 cells labeled by six observed cell types derived from the gene expression measurements: endoderm (233 cells), endothelial (124 cells), primordial germ cell-like cell (PGCLC; 233 cells), somite (3,188 cells), neural tube (2,289 cells) and NMP (513 cells) (Methods).

a, A representative live-imaged TLS at 120 hours with NMP pool (orange) and elongating neural tube (light blue) and somite (green) structures. A total of 14 TLSs were generated. b, Normalized discrepancy \(\tilde{D}({\mathcal{T}},{F}_{S})\) of cell differentiation maps, inferred by Carta-Tree (blue) and Carta-DAG (orange), for increasing number k of progenitors revealing the Pareto fronts. Discrepancy of cell differentiation maps inferred by existing methods that infer unobserved progenitors (⋅) and do not infer unobserved progenitors (X) are also shown. c, Canonical model of TLS differentiation29. The included cell types are notated as follows: M, neuromesodermal progenitor (NMP); N, neural tube; S, somite; T, endothelial; D, endoderm; P, primordial germ-cell-like cell (PGCLC). d, Cell differentiation map inferred by Carta-DAG, where edges are annotated by number of cells that traverse the cell type transition. Legend indicates the number of cells of each cell type. e, Cell differentiation map inferred by PhyloVelo. Weight of each edge is the inferred transition probability between two cell types. f, The number of cells that directly transition from progenitor cell types (rows) to observed cell types (columns) for the cell differentiation maps inferred by each method. g, The marginal distribution, or the proportion of total cells that derive from each progenitor, and the corresponding entropy H of each distribution. h, The support, or the number of ancestral cells in the cell lineage trees that have an observed potency that exactly matches the potency of an inferred progenitor.
We compared the cell differentiation maps generated by Carta with varying numbers of progenitors (Extended Data Fig. 1) to the differentiation maps inferred by existing methods. Both modes of Carta—that is, Carta-Tree and Carta-DAG—consistently infer cell differentiation maps with lower discrepancy compared to existing methods for the same number of progenitors (Fig. 3b). For example, the normalized discrepancies of cell differentiation maps with k = 5 progenitors that Carta-Tree and Carta-DAG infer are 0.802 and 0.668, respectively. By contrast, ICE-FASE, EvoC and Fitch infer cell differentiation maps that have five progenitors and normalized discrepancy of 1.936, 2.580 and 0.915, respectively (Methods). PhyloVelo infers a map with six progenitors with normalized discrepancy of 1.930 compared to 0.802 and 0.546 for Carta-Tree and Carta-DAG, respectively, with k = 6. We determined the optimal number k* = 7 progenitors in the cell differentiation map (normalized discrepancy 0.458) by identifying the ‘elbow’ in the Pareto fronts derived using Carta-DAG (Methods).
The cell differentiation map inferred by Carta (Fig. 3d) agrees with known features of trunk developmental progression. Carta-Tree infers a cell differentiation map in which the relative ordering of commitment of observed cell types is PGCLC, endoderm, endothelial, NMP, somites and neural tube (Extended Data Fig. 2). This is consistent with the canonical model of TLS differentiation in which the fate of PGCLC and endoderm cells is committed earlier compared to the NMP, somite and neural tube cells (Fig. 3c)48. This is also reflected in the Carta-DAG cell differentiation map, in which endothelial, endoderm and PGCLC cells derive from progenitors with larger potencies (mean progenitor potency size: 4.5, 4.0 and 5.0, respectively) compared to the more closely related NMP, somitic and neural tube cells that arise from progenitors with more restrictive potencies (mean progenitor potency size: 3.7, 3.3 and 3.3, respectively). We demonstrate that the inferred cell differentiations maps are robust to low sampling counts of cells in the dataset (Supplementary Section 9 and Supplementary Figs. 8 and 9).
A key insight of the Carta-DAG cell differentiation map is the convergent differentiation of somite cells, with one origin stemming from shared ancestry with neural tube cells and an alternate origin indicating shared ancestry with endothelial cells via the presence of the {endothelial, somite} progenitor. This is consistent with previous in vivo studies that found evidence for a secondary pathway toward the production of the trunk endothelium58,59. Such instances of convergent differentiation cannot be revealed by methods such as ICE-FASE and EvoC that infer only tree-structured cell differentiation maps in which each cell type arises from a single developmental trajectory.
Carta further reveals the progenitor dynamics as well as the commitment bias of NMPs—that is, the proportion of NMPs committing to each downstream state. The Carta-DAG differentiation map includes NMPs in multiple known stages of development55,56,57. The {NMP} cell type represents observed undifferentiated NMPs; the {neural tube, somite} cell type represents ancestral NMPs that existed in the past; and the {NMP, neural tube, somite} cell type represents NMP cells that both self-renew and are differentiating. Furthermore, the {NMP, neural tube} cell type represents NMP cells that are only observed differentiating into neural tube cells, and the {NMP, somite} cell type represents NMP cells that are only observed differentiating into somitic cells. Notably, all of these different instances of progenitor cell types can be represented simultaneously only in a DAG structure and not in a tree structure. We observe that the Carta-DAG cell differentiation map only includes the {NMP, neural tube} and not the {NMP, somite} progenitor, suggesting that NMP cells in this system have a higher propensity to commit to a neural rather than a somitic fate (Fig. 3d). This bias toward neural fate supports previous analyses that NMP cells gradually shift their differentiation potential toward the neural fate during TLS development29.
By contrast, methods where all progenitor cell types are assumed to be observed, such as Fitch and PhyloVelo, infer cell differentiation maps that are not well supported by the literature. Many spurious cell type transitions exist in the differentiation map produced by PhyloVelo (Fig. 3e). For example, somitic cells differentiate into PGCLC, endoderm and neural tube cells. Furthermore, endothelial cells differentiate into somites, and endoderm cells differentiate into NMPs. In these instances, observed cell types are shown to transition directly to each other when it is known that these cell types are related through progenitor cell types that are potent for each of them. This highlights the deficiencies of the assumption that all progenitors are observed. Additionally, PhyloVelo does not correctly infer the hierarchical differentiation process, as the differentiation map shows that neural tube cells can differentiate back to the NMP state (Fig. 3e). The cell differentiation maps inferred by ICE-FASE, EvoC and Fitch also have poor agreement with the reported developmental routes in TLSs (Extended Data Fig. 2).
The progenitors inferred by Carta are better supported by the cell types of descendants of ancestral cells in the cell lineage trees compared to existing methods. We demonstrate this advantage using two metrics. First, we calculate the distribution of the number of cells of each observed cell type that directly arise from the progenitor cell type inferred by each method (Fig. 3f and Methods). The progenitors inferred by Carta produce a more uniform distribution (Fig. 3g) quantified by the higher entropy (H = 1.759) of the distribution compared to existing methods (ICE-FASE: H = 1.0, EvoC: H = 0.820, PhyloVelo: H = 1.104, Fitch: H = 1.319). Moreover, for ICE-FASE, EvoC and PhyloVelo, the proportion of cells arising from the two progenitors that account for the most cells (ICE-FASE: 0.814, EvoC: 0.954, PhyloVelo: 0.859, Fitch: 0.719) is substantially larger than the proportion of 0.525 for Carta. Second, we calculate the ‘support’ of a set of inferred progenitors—that is, the number of ancestral cells in the cell lineage trees that have an observed potency that matches the potency of an inferred progenitor in that set (Fig. 3h). Progenitors inferred by Carta have much higher support C = 1,306 compared to existing methods (ICE-FASE: C = 382, EvoC: C = 41, PhyloVelo: C = 360, Fitch: C = 459), indicating that Carta differentiation map provides a better fit with the input cell lineage trees.
Carta reveals mouse hematopoiesis progenitor hierarchy
We applied Carta and several existing methods to a single-cell lineage tracing dataset of mouse hematopoiesis30 and compared the resulting cell differentiation maps. This dataset was obtained by inserting random and heritable lentiviral barcodes in mouse hematopoietic stem cells (HSCs), which were then allowed to differentiate in vitro, with the culture sampled at days 2, 4 and 6. scRNA-seq was performed to simultaneously measure the barcodes and gene expression of sampled cells, with barcodes being captured for 49,302 of these cells. The barcode measurements were used to construct 5,864 star-shaped cell lineage trees, one for each unique barcode shared across multiple cells (Methods). This is in contrast to CRISPR–Cas9-based lineage tracing data on TLSs that have far fewer trees (14) but with higher depth (median, 9). We label cells into nine cell types following the annotation from Weinreb et al.30—megakaryoctyes (1,035 cells), erythrocytes (365 cells), mast cells (1,414 cells), basophils (5,514 cells), eosinophils (168 cells), neutrophils (8,555 cells), monocytes (8,165 cells), dendritic cells (including plasmocytoid dendritic cells (pDCs) and Ccr7+ migratory dendritic cells (migDCs); 113 cells) and lymphoid cells (203 cells)—with the remaining cells marked as undifferentiated (23,770 cells) (Fig. 4a). Note that more recent studies split dendritic cells into two major subpopulations, classical/conventional dendritic cells (cDCs) and pDCs, but this dataset does not contain cDCs60,61,62.

a, Low-dimensional visualization83 of scRNA-seq of 43,670 clonally barcoded cells in varying stages of mouse hematopoesis differentiation30. Cells are colored by cell type, and legend contains the number of cells of each cell type. b, Normalized discrepancy \(\tilde{D}({\mathcal{T}},{F}_{S})\) of cell differentiation maps inferred by Carta and existing methods with varying numbers of progenitors. c, Progenitors and their proportions inferred by Carta with increasing number k of progenitors. d, Canonical model of the hierarchy of progenitors during mouse hematopoiesis10,64,65,66,67,68. Dashed arrows show alternate routes of differentiation that were suggested in previous studies. e, Cell differentiation map inferred by Carta (e) and distance-based heuristic employed by Weinreb et al.30 (f). Red indicates inferred progenitors that are non-canonical—that is, do not agree with the canonical model shown in d. GMP, granulocyte-monocyte progenitors; MDP, monocyte-dendritic progenitors; EP, erythrocyte progenitor; GP, granulocyte progenitor.
We compared the differentiation maps inferred by both modes of Carta, Carta-Tree and Carta-DAG (runtimes: Supplementary Table 2) to cell differentiation maps published in the original study (Weinreb et al.30) and inferred using existing methods: Fitch63, PhyloVelo54, ICE-FASE47 and EvoC39 (Fig. 4b and Methods). PhyloVelo infers a cell differentiation map with only five progenitors but much higher normalized discrepancy of 1.809, whereas Fitch infers a map with nine progenitors with low normalized discrepancy of 0.186. By contrast, Carta-DAG cell differentiation maps with the same number of progenitors have much lower discrepancies (0.4 for five progenitors and 0.039 for nine progenitors). Similarly, ICE-FASE, EvoC and Weinreb et al.30 infer tree-structured cell differentiation maps comprising eight progenitors but with higher normalized discrepancy of 0.921, 3.598 and 2.51, respectively, compared to Carta-Tree with the k = 8 progenitors and normalized discrepancy of 0.738. Because the canonical model of hematopoiesis64 is tree structured (Fig. 4d), we focus our attention on the cell differentiation maps inferred by Carta-Tree with k = 1–10 progenitors. As the number k of progenitors is increased, Carta-Tree reveals a more fine-grained structure of differentiation, with several inferred progenitors shared across the cell differentiation maps (Fig. 4c and Extended Data Fig. 3). Specifically, Carta-Tree infers only nine distinct progenitors across the 10 cell differentiation maps, with the results for k = 8, 9 and 10 having the same set of progenitors. We show that the inferred Carta-Tree cell differentiation maps are robust to different cell type definitions and to subsampling of the cells in the data (Supplementary Sections 9 and 10 and Supplementary Figs. 8 and 10). We use a heuristic to obtain the elbow of the Pareto front (Methods) and identify an optimal Carta-Tree cell differentiation map with k* = 7 progenitors and normalized discrepancy of 0.762.
The cell differentiation map inferred by Carta aligns more closely with the canonical model of hematopoiesis64 compared to the hierarchy of progenitors published in the original study30. Weinreb et al. employed a heuristic algorithm similar to EvoC28, which involves hierarchical clustering of the cell types based on a measure of clonal coupling (proportional to the number of shared lineage barcodes) to construct the lineage hierarchy. We compared the Carta cell differentiation map (Fig. 4e) and the lineage hierarchy generated by Weinreb et al. (Fig. 4f) to the canonical model of murine hematopoiesis64 (Fig. 4d). Carta infers that the myeloid cells (mast, basophil, eosinophil, neutrophil and Mo) originate from a common unobserved progenitor cell type, which is consistent with the common myeloid progenitor (CMP) in the canonical model of hematopoiesis10,64,65,66,67,68. Carta also infers an intermediate non-canonical progenitor, which we identify as myeloblast69, with potency for basophil, eosinophil, neutrophil and monocyte cells. By contrast, Weinreb et al.30 suggest that the myeloid cells separate into two trajectories (first containing mast, basophil and eosinophil and second containing neutrophil and monocyte) very early during differentiation when the cells are still multipotent progenitors (MPPs). Carta additionally identifies an unobserved progenitor restricted to megakaryocytes and eythrocytes, known as the megakaryocyte–erythrocyte progenitor (MEP), which arises directly from MPP cells. This finding is consistent with previous studies that found evidence that, in mouse, MPP gives rise to MEP without passing through the CMP64,70,71,72,73. Although Weinreb et al.30 also identify MEP, they propose that it originates from a non-canonical progenitor that is potent for megakaryocytes, eythrocytes and mast cells. Carta also correctly infers that lymphoid and dendritic cells belong to a differentiation trajectory that separates early from the other cell types (myeloids, megakaryocytes and erythrocytes) during hematopoiesis64,71. However, it does not identify the presence of the common lymphoid progenitor (CLP), possibly due to low sampling of lymphoid and dendritic cells (18 and 22 cells, respectively) in the data (Supplementary Section 11 and Supplementary Fig. 11). By contrast, Weinreb et al.30 identify the CLP but suggest that it originates from a non-canonical hierarchy of progenitors with potency for neutrophils and monocytes. The Carta cell differentiation tree has the lowest Robinson–Foulds distance74 with the canonical tree (1; maximum possible, 7), compared to the tree inferred by Weinreb et al. (4; maximum possible, 8), the ICE-FASE tree (6; maximum possible, 8) and the EvoC tree (2; maximum possible, 8) (Extended Data Fig. 4).
We examine the concordance between the progenitor cell types of undifferentiated cells predicted by Carta and the gene expression of these cells. Specifically, we determined the progenitor cell type of undifferentiated cells sampled at day 2 based on the potency of their ancestors in the cell lineage trees inferred by Carta. We found that the undifferentiated cells have similar gene expression to the observed cell types in the potency set—that is, the cell types that Carta predicts the undifferentiated cell will differentiate into (Fig. 5a and Extended Data Fig. 5). We quantify this similarity by comparing the predicted fate of undifferentiated cells to the cell type cluster of the closest cell in gene expression space (Methods). We observe a high degree of overlap between predicted potency and closest mature cell type in the cases where the inferred progenitor is potent for that cell type (Fig. 5b,c). Because Carta uses only lineage information and not gene expression in inferring progenitors, these results provide orthogonal validation for the progenitor cell types inferred by Carta.

a, Progenitor predictions given by Carta (colored dots) for undifferentiated cells sampled at day 2. b, Potencies of the inferred progenitor cell types. c, The proportion of undifferentiated cells (from days 2, 4 and 6) that are closest in transcriptional space to the indicated observed cell type for each predicted progenitor. Ba, basophil; Eo, eosinophil; Mo, monocyte; Neu, neutrophil.
Discussion
We introduce Carta to infer cell differentiation maps from cell lineage trees while accounting for sampling limitations in high-throughput lineage tracing data and inherent biological variability in development. Carta employs a mathematical model of a cell differentiation map in which a progenitor cell type is defined by its potency—that is, the set of cell types that can be attained by their descendants. This model enables the inference of transient progenitor cell types that arise during development but may not be observed in the lineage tracing data. A key insight of our work is that there exists a tradeoff between the number of progenitors in the cell differentiation map (a measure of its ‘complexity’) and how well the map fits the input cell lineage trees (‘discrepancy’). Carta explicitly evaluates this tradeoff by computing the Pareto front of cell differentiation maps and selecting a map with an optimal number of progenitor cell types.
Carta has several limitations and presents multiple opportunities for future development. First, Carta takes cell lineage trees derived from lineage tracing data as input, but these trees are not always accurate44. Joint inference of a cell lineage tree and a cell differentiation map from lineage tracing data in a multi-objective optimization framework might yield more accurate trees and maps. Second, our discrepancy measure is based on maximum parsimony and counts the number of unobserved descendants of labeled progenitors. Extending this score to a probabilistic model for cell differentiation and fate commitment during development is a promising future direction with some relation to the structure learning problem in probabilistic graphical models75. Third, Carta quantifies the complexity of the differentiation map by the number of progenitors, but complexity could also be described in terms of the number and type of transitions (Supplementary Section 1). Finally, Carta assumes that progenitors do not regain potency for a cell type after differentiation. Although this assumption is reasonable for most normal developmental systems, dedifferentiation does occur in aberrant systems such as cancer and is a major mechanism of cancer progression, cancer cell plasticity and immune evasion76,77,78,79. Extending Carta to model dedifferentiation would be useful for modeling cancer development or stem cell reprogramming.
Finally, investigations of developmental systems are increasingly using other high-throughput technologies, such spatial transcriptomics31,80,81 and single-cell multimodal sequencing82. Combining lineage tracing with spatial and/or multimodal single-cell sequencing is crucial for measuring the interplay among microenvironment, epigenetic regulation and lineage of the cells. We envision that Carta will play a crucial role in distinguishing the relative contributions of cell lineage, cell differentiation and spatial location during development and provide a foundation for future development of algorithms for cell differentiation mapping.
Methods
Definition and inference of cell differentiation maps
A ‘cell differentiation map’ FS describes the differentiation of cells into observed cell types S. Here, we give a formal definition of a cell differentiation map FS and formulate the problem of inferring a cell differentiation map from a set \({\mathcal{T}}\) of cell lineage trees.
We define a cell differentiation map FS to be a vertex-labeled directed graph whose sinks—that is, vertices with outdegree d = 0—are the observed cell types S, and whose internal vertices—that is, vertices with outdegree d > 0—are the progenitor cell types. The directed edges of FS describe the cell type transitions that occurred during development. Each sink vertex (observed cell type) t ∈ S is labeled by the singleton set {t} (or, for simplicity, by an element of S), and each internal vertex (progenitor cell type) is labeled by a potency set—that is, a subset of S.
We model development as a process in which cells progressively lose potency and do not regain potency for a cell type once it is lost. Thus, FS is a DAG—that is, does not have directed cycles—in which the root of FS has label S indicating the totipotent cell with potency S, and the internal vertices have unique labels that satisfy the following two conditions. First, because we assume that cells lose potency only during development, every directed edge \((P,{P}^{{\prime} })\) in FS satisfies \({P}^{{\prime} }\subset P\). Second, by definition of potency, for each cell type t ∈ S, there exists a directed path in FS from a progenitor P to an observed cell type {t} if and only if it is potent for the cell type t—that is, t ∈ P. Consequentially, the vertex set \({{\mathcal{P}}}_{S}\) of a cell differentiation map FS always contains the totipotent cell S and the singleton set {t} for each observed cell type t ∈ S.
The cell types of ancestral cells are determined by a labeling of the internal vertices of \({\mathcal{T}}\) (ancestral cells) by the vertices of the cell differentiation map FS (cell types). Such a labeling must be compatible with the trees \({\mathcal{T}}\) and cell differentiation map FS—that is, it must satisfy the following two conditions. First, each ancestral cell in a cell lineage tree must be labeled by a potency that contains all the observed cell types of its descendants in the tree. Second, cell type transitions determined by the labeling—that is, edges in the lineage trees connecting vertices labeled by distinct cell types—must be supported by the cell differentiation map FS. More formally, for every edge (u, v) in a cell lineage tree, there must exist a path from ℓ(u) to ℓ(v) in FS.
For cell lineage trees \({\mathcal{T}}\) and a cell differentiation map FS, there may be multiple compatible labelings. We evaluate a labeling ℓ by its discrepancy, defined as the number of instances when a cell type in the potency ℓ(v) of an ancestral cell v is not observed in its descendants—that is, the leaves of the subtree rooted at v. More formally,
where \({\mathbb{1}}\) is the indicator function and B(v) is the set of observed cell types of the descendants of cell v.
We define the discrepancy between the cell lineage tree \({\mathcal{T}}\) and a cell differentiation map FS by the minimum discrepancy obtained over all compatible labelings—that is,
where \({\mathcal{C}}({\mathcal{T}},{F}_{S})\) is the set of compatible labelings for cell lineage trees \({\mathcal{T}}\) and cell differentiation map FS. A more general description of discrepancy is given in Supplementary Section 1.
As such, evaluating the discrepancy of a given cell differentiation map FS with a set of cell lineage trees \({\mathcal{T}}\) is equivalent to finding a compatible vertex labeling ℓ that minimizes the induced discrepancy \(D({\mathcal{T}},{F}_{S},\ell )\). We refer to this as the Progenitor Labeling Problem (PLP) and formally pose it as follows.
Problem: Progenitor Labeling Problem
Given a set \({\mathcal{T}}\) of cell lineage trees and cell differentiation map FS, find a valid labeling ℓ that minimizes the discrepancy \(D({\mathcal{T}},{F}_{S},\ell )\).
This is an analog of the ‘small parsimony’ problem63, and we show that it can be solved by a dynamic program by adapting Sankoff’s algorithm84 (Supplementary Section 3). In practice, we only observe the cell lineage trees \({\mathcal{T}}\) and must infer the cell differentiation map FS. Due to technical limitations in current lineage tracing technologies, such as limited sampling of cells, inferring a map FS with minimum discrepancy \(D({\mathcal{T}},{F}_{S})\) may lead to a large number of progenitors, many of which may be false positives. As such, we pose the cell differentiation map inference problem (CDMIP) of inferring a cell differentiation map with minimum discrepancy for a fixed number k of progenitors.
Problem: Cell Differentiation Map Inference Problem
Given cell lineage trees \({\mathcal{T}}\) with observed cell types S and integer k, find a cell differentiation map FS with k progenitors such that \(D({\mathcal{T}},{F}_{S})\) is minimized.
An interesting special case of the CDMIP is when the differentiation map is restricted to be a tree. We define this problem as follows.
Problem: Cell Differentiation Tree Inference Problem
Given cell lineage trees \({\mathcal{T}}\) with observed cell types S and integer k, find a cell differentiation tree FS with k progenitors such that \(D({\mathcal{T}},{F}_{S})\) is minimized.
We show that both the CDMIP and the Cell Differentiation Tree Inference Problem (CDTIP) are nondeterministic polynomial-time hard (NP-hard) (see Supplementary Sections 4 and 5 for the characterization of the solutions and Supplementary Section 6 for the complexity proofs).
Carta: an algorithm for cell differentiation mapping
We developed Carta, an algorithm to infer a cell differentiation map FS from cell lineage trees \({\mathcal{T}}\) that balances the tradeoff between the discrepancy \(D({\mathcal{T}},{F}_{S})\) and the number k of progenitors in the cell differentiation maps. Carta allows inference of DAG and tree-structured cell differentiation maps by solving this multi-objective optimization problem in two steps, which we detail below.
First, Carta finds the cell differentiation map with minimum discrepancy for each number k of progenitors across a range of values of k. This reveals the Pareto front, indicating the minimum discrepancy obtained over differentiation maps for each fixed number k of progenitors. Carta has two modes: Carta-DAG to generate DAG-structured cell differentiation maps and Carta-Tree to generate tree-structured cell differentiation maps. For a fixed number k of progenitors, Carta-DAG and Carta-Tree use mixed integer linear programming (MILP) to solve the CDMIP and the CDTIP, respectively. Second, Carta determines the optimal number k* of progenitors by identifying the elbow of the Pareto front. To this end, we use ‘kneedle’85, a heuristic algorithm that finds the point of maximum curvature on the Pareto front. The edges of the cell differentiation map are determined by including all cell type transitions that appear frequently in the labeled cell lineage trees.
Carta also includes a heuristic that takes as input a set of putative progenitors to be supplied by the user. In this mode, Carta infers a cell differentiation map that has k of the progenitors from the list supplied by the user such that discrepancy is minimized. Carta employs a different MILP formulation (Supplementary Section 2) in which the number of binary variables depends only on the size of the input progenitor set and, thus, is scalable to large values of the number k of progenitors, the number of observed cell types and the size of cell lineage trees. The MILPs are solved using the Gurobi Optimizer86 in Python, and the details of the MILP formulations are described in Supplementary Section 2.
Simulation details
Simulating cell differentiation maps
Here, we describe our procedure to obtain and generate tree-structured and DAG-structured cell differentiation maps that define the ground truth differentiation process in our simulations.
Binary-tree-structured cell differentiation maps: We selected 20 binary-tree-structured cell differentiation maps with 16 observed cell types generated by Fang et al.47—10 from the ‘balanced-TBR’ and 10 from the ‘random’ class. The ‘balanced-TBR’ class consists of cell differentiation maps that are generated by applying one random tree bisection and reconnection (TBR) to a fully balanced binary tree. The ‘random’ class delineates cell differentiation maps that are generated by a random sequence of recursive bifurcations. From each selected cell differentiation map, we additionally generate binary tree cell differentiation maps with eight and 12 observed cell types by sampling eight and 12 observed cell types uniformly at random and pruning the observed cell types that are not sampled. The final set of binary-tree-structured cell differentiation maps contains 20 maps of each of 8, 12 and 16 observed cell types.
Tree-structured cell differentiation maps with polytomies: For each of the 20 binary tree cell differentiation maps, we introduce two polytomies—that is, a node with more than two children—by randomly choosing a progenitor node with at least one child node that is also a progenitor and contracting it. In the contraction for a node, we remove all child nodes that are progenitors and create directed edges between the chosen node and the children of the progenitor child nodes. This process results in 20 non-binary-tree-structured differentiation maps.
DAG-structured cell differentiation maps: We generated DAG-structured cell differentiation maps by merging nodes and contracting edges in each binary-tree-structured differentiation map as follows. We sampled two disjoint pairs of observed cell types (leaves of the cell differentiation maps) of the tree-structured differentiation maps uniformly at random and merged each pair. This generates two instances of convergent differentiation—that is, an event in which differing developmental trajectories converge toward the same cell type. We also introduced a polytomy in each DAG-structured differentiation map by randomly sampling a progenitor whose children are both progenitors and contracting its outgoing edges in the same manner as in the trees with polytomies case. The resulting DAG-structured differentiation maps have k = m − 1 progenitors, where m is the number of observed cell types. Using this procedure, we generate 20 DAG-structured cell differentiation maps of each of 6, 10 and 14 observed cell types from binary-tree-structured cell differentiation maps with 8, 12 and 16 observed cell types, respectively.
Simulating cell lineage trees from a cell differentiation map
For each simulated cell differentiation map FS, we simulated time-resolved binary cell lineage trees that follow the differentiation routes specified by that map. To generate tree topologies, we used the generalized, forward time birth–death simulator included in the Cassiopeia platform40. Let z be the number of cells sampled per extant cell type, and let α be the subsampling rate. The process terminates when \(\frac{| S| * z}{\alpha }\) extant tips are sampled. We drew birth waiting times from a shifted exponential distribution with a shift constant of c = 0.01 and estimated the birth and death rates to produce trees with total times of approximately 1 for the given number of extant tips. We then normalized the branch lengths of T such that the longest path from the root to one of a leaf of the tree is of length 1 to match the times on FS.
We simulated cell type differentiation in two steps. First, we assigned a differentiation time for each cell type transition in the cell differentiation map. Specifically, we annotated each vertex of the cell differentiation map by a time between 0 and 1, representing the time of arrival of that cell type such that if vertex u precedes vertex v, then τ(u) < τ(v). These times are determined by a process in which we iterated through paths in the cell differentiation map from root to sink and, on each iteration, annotated the length of each edge in a path by evenly splitting the remaining length of that path among its edges. The time of each vertex is the sum of the path length from the root. Second, we randomly labeled the ancestral cells of each cell lineage tree T with cell types such that cell type transitions in T are consistent with the cell type transitions in the cell differentiation map FS. To achieve this, we first initialized the label ℓ(r(T)) of the root vertex r of cell lineage tree T as the totipotent progenitor S. Let τT and \({\tau }_{{F}_{S}}\) be the timepoint annotation function for the cell lineage tree T and cell differentiation map FS, respectively. We performed a depth-first, preorder traversal of the edges (u, v) ∈ E(T) of the lineage tree such that we annotate ℓ(v) as ℓ(u) if \({\tau }_{T}(v) > {\tau }_{{F}_{S}}(\ell (u))\) and otherwise a randomly sampled descendant of ℓ(u) in the FS. Finally, once each cell in the cell lineage is annotated with a progenitor label, we randomly sampled the specified number z = 50, 100 or 200 of cells labeled with each extant cell type in S. We took the subtree induced by the sampled cells as well as the cell type labelings of the leaves of this tree as the final inputs to our cell differentiation map inference algorithms.
We note that a set of cell lineage trees are provided by Fang et al.47 that accompany their binary tree cell differentiation maps. These trees are generated through a coalescent process that models both cell division and differentiation simultaneously. As these cell differentiation maps correspond only to binary tree cell differentiation maps, we choose to simulate our own cell lineage trees for non-binary tree and DAG cell differentiation map structures. We additionally show that the simulated cell lineages in Fang et al. may not be suitable for our evaluation due to the lack of asymmetric cell division in their simulation process (Supplementary Section 7).
Simulating CRISPR–Cas9 timed cell lineage trees
We simulate CRISPR–Cas9 mutagenesis data similarly to that of recent lineage tracing experiments28,29,38,39,40 using the ‘Cas9LineageTracingDataSimulator’ class in the Cassiopeia package. We set parameters that reflect observations from existing lineage tracing datasets28,49: 30 editable sites, 50 observed indel outcomes per site, 40% probability of an edit appearing at a cell at a site and 25% missing data. We then used the ‘IDEExponentialMLE’ function that implements ConvexML87 to estimate branch lengths on the cell lineage tree.
Implementation details for various methods in simulated data
Here, we provide additional details about how Carta as well as ICE-FASE and EvoC are run on simulations.
We run Carta-DAG with k = k* − 3, …, k* + 3, where k* is the true number of progenitors, and then use the kneedle algorithm85 to select the optimal number of progenitors in the cell differentiation map. We also run Carta-Tree in a similar manner when the ground truth cell differentiation map is a tree with polytomies. Following the assumption in ICE-FASE and EvoC, which always generate cell differentiation maps with exactly ∣S∣ − 1 progenitors, we run Carta-Tree with k = ∣S∣ − 1 cell types when the ground truth cell differentiation map is a DAG or when it is a binary tree. ICE-FASE and EvoC are applied with default settings to each single timed cell lineage tree with cell type annotation on the leaves.
We apply the heuristic mode of Carta in our simulations. For simulated cell lineage trees in which the ground truth cell differentiation map has ∣S∣ > 6 observed cell types, we provide the set of unique observed potencies of the ancestral cells in the cell lineage tree as a set of putative progenitors. This set is guaranteed to include all ground truth progenitors assuming that at least one ancestral cell with each progenitor label gives rise to all observed cell types for which it is potent. Furthermore, this heuristic leads to efficient scaling with the number of observed cell types, the number of cells in the cell lineage tree (Supplementary Fig. 6) and the number of progenitors in the inferred cell differentiation map (Supplementary Fig. 7 and Supplementary Tables 1 and 2). For simulated cell lineage trees in which the ground truth cell differentiation map has ∣S∣ = 6 observed cell types, we provide the full set of 57 possible progenitors.
Simulation metrics
We evaluate the inferred cell differentiation maps against the simulated ground truth cell differentiation maps using two metrics that quantify the difference in the progenitors in each:
(1) Jaccard distance \({d}_{J}({\mathcal{P}},{{\mathcal{P}}}^{* })\) (ref. 53):
where \({{\mathcal{P}}}^{* }\) and \({\mathcal{P}}\) are the ground truth and the inferred set of progenitors, respectively.
The Jaccard distance \({d}_{J}({\mathcal{P}},{{\mathcal{P}}}^{* })\) is 0 if and only if the set \({\mathcal{P}}\) of inferred progenitors exactly matches the set \({{\mathcal{P}}}^{* }\) of ground truth progenitors.
(2) The normalized minimum Hamming distance \({d}_{H}({\mathcal{P}},{{\mathcal{P}}}^{* })\):
where Hamming distance88 dH(P*, P) = ∣P*\P∣ + ∣P\P*∣.
Intuitively, the Hamming distance of two progenitors is defined as the size of the symmetric difference of the two progenitors and would be 0 if and only if the two progenitors are identical. The normalized minimum Hamming distance is the sum of the minimum Hamming distance between an inferred progenitor and all of the progenitors in the ground truth, normalized by the number \(| {{\mathcal{P}}}^{* }|\) of ground truth progenitors and number ∣S∣ of observed cell types. As such, \({d}_{H}({\mathcal{P}},{{\mathcal{P}}}^{* })\) is 0 if and only if each ground truth progenitor is present in the inferred set \({\mathcal{P}}\) of progenitors.
We further evaluate the precision and recall of the progenitors inferred by each method against the ground truth, where the precision is defined as:
and the recall is defined as:
Data processing details
Processing of TLS data
We obtained 14 cell lineage trees that record the cell division of 14 TLSs generated in ref. 29. These lineages were generated using scRNA-seq readout from mouse embryonic stem cells engineered with CRISPR–Cas9 lineage tracing technology. These scRNA-seq data were then input to the Cassiopeia lineage preprocessing and reconstruction package40. The branch lengths are not given by Cassiopeia, and, hence, we used unit branch lengths. Each observed cell (leaf) in each cell lineage tree was assigned a cell type by a previously published ref. 48. We grouped all somite cell subtypes (somite (−1), somite 0, somite, sclerotome-like and dermomyotome-like) into one umbrella type ‘somite’, and we grouped NeuralTube1 and NeuralTube2 cell types into one umbrella type ‘neural tube’. We then pruned from our trees each leaf labeled with a cell type not included in our analysis (anterior presomitic mesoderm, posterior presomitic mesoderm).
As a preprocessing step to Carta only, we collapsed each clade in each cell lineage tree comprising extant cells that share a cell type into a single extant cell with that cell type. These clades do not contribute to cell type transitions nor the objective score of Carta.
Processing of data from Weinreb et al. study
We obtained the in vitro differentiation timecourse data generated by Weinreb et al.30 from their public repository (https://github.com/AllonKleinLab/paper-data/tree/master/Lineage_tracing_on_transcriptional_landscapes_links_state_to_fate_during_differentiation). The associated metadata include the lentiviral barcode and cell type of each cell. Each of the 5,864 barcodes corresponds to a star-shaped cell lineage tree, where the leaves represent the sequenced cells that contain that barcode and are annotated by cell types. Of the 130,887 cells in the dataset, 49,302 have an associated barcode. We observed 107 distinct observed potencies in the data, defined by the set of cell types of the descendants of a cell, even though the data have only nine extant cell types. This is possibly due to cell sampling limitations, as discussed in the ‘Statistics and reproducibility’ section. As such, we performed a mild filtering of the data by removing barcodes in which the observed potency occurs fewer than 10 times in the data. This step removes only 4.1% of the barcodes, resulting in 5,642 cell lineage trees totaling 43,670 cells. We merged the ‘pDC’ and ‘Ccr7 DC’ cell types into one ‘DC’ cell type and removed cells with the undifferentiated cell type from the cell lineage trees. These cell lineage trees are provided as input for Carta and the other existing methods.
Implementation and application of existing methods
Fitch’s algorithm
Fitch’s algorithm solves the small parsimony problem63, which can be applied to lineage tracing data to build cell differentiation maps under the assumption that all the progenitor cell types are observed in the data. In brief, given a phylogeny with each leaf labeled with one of a set of states, the small parsimony problem seeks to find the labeling of internal nodes of a phylogeny with those states such that the fewest number of transitions in state between parent and child nodes is obtained63. The frequency of transition from cell type i to cell type j can then be counted as the number of transitions from an internal cell labeled i to one labeled j in this labeled phylogeny.
For the dataset from Weinreb et al., we directly applied Fitch’s algorithm and totaled the number of transitions between cell types across the Fitch labeling for each star-shaped cell lineage tree. We then stored these totals in a cell type transition matrix and row normalized the matrix, converting transition frequencies to transition proportions that sum to 1 for each cell type of origin.
For the TLS dataset, to account for the often large number of equally parsimonious Fitch labelings for large trees, we used FitchCount38, which efficiently counts the total number of transitions between cell types in all equally minimal Fitch labelings. As the total number of transitions counted by FitchCount increases rapidly by the size of the cell lineage tree, the transition counts on large trees would dominate the transition count totaled over all trees. Thus, we computed a normalized sum of the transitions over all trees. For each tree, we generated a row-normalized cell type transition matrix from the FitchCount transitions and then computed the sum of these matrices as the final cell type transition matrix. This final matrix is then row normalized.
EvoC
EvoC is defined as the normalized phylogenetic distance between any pair of cell annotations on a tree39. We extend the definition given in Yang et al.39 to multiple trees \({\mathcal{T}}\). Given cell types M and K, the average phylogenetic distance between leaves (extant cells) labeled by these cell types on the cell lineage tree is defined as:
where \({\{m,k\}}_{{T}_{i}}\) is the set of all pairwise combinations of leaves with type M and K on tree Ti, and \({d}_{{T}_{i}}(i,j)\) denotes the phylogenetic distance between leaves on tree Ti. Intuitively, this metric calculates the average phylogenetic distance between two cells of cell types M and K. We then perform hierarchical clustering on the cell types based on Devo using the unweighted pair group method with arithmetic mean (UPGMA) algorithm89, yielding a tree structure cell differentiation map.
ICE-FASE
ICE-FASE calculates the average times at which cell types separate across given time-resolved cell lineage trees and performs hierarchical clustering between these cell types to form the resultant cell differentiation map47. To run ICE-FASE, we used the implementation in the QFM package in R47. In addition to cell type annotations, ICE-FASE requires time-resolved phylogenies with branch lengths as input. For the TLS cell lineage trees, we estimated the branch lengths using the Maximum Likelihood Branch Length Estimator implemented in Cassiopeia40. The lineage tracing data from Weinreb et al.30 are already annotated with time.
We implemented several workarounds in the analysis of both datasets owing to limitations in the ICE-FASE codebase. First, because the ICE-FASE code crashes when multiple trees are given as input, we created a single tree by connecting the root of each input tree by a 0-length branch to a dummy root node. Second, the ICE-FASE code is not equipped to handle trees that have polytomies—that is, vertices with more than two children. Because the TLS trees and the Weinreb et al. trees both have such polytomies, we arbitrarily binarized these trees by creating edges with 0 length. Notably, because ICE-FASE depends only on the timing at which cells separate, the introduction of these 0-length branches should not affect the analysis. Moreover, combining multiple trees into a single tree should not be problematic, as all pairs of cells in different trees now connected by the dummy root have a separation time of 0.
PhyloVelo
PhyloVelo attempts to learn the differentiation trajectories of a system from gene expression data that are informed by the lineage depth of each cell. To run PhyloVelo, we used the PhyloVelo package as provided in ref. 54. We performed the analysis very closely to the analysis of PhyloVelo performed in that study. For both datasets, we used the ‘velocity_inference’ and ‘velocity_embedding’ embedding functions to calculate the PhyloVelo trajectories and then passed the output of these functions to the ‘state_graph’ function in Dynamo90 to obtain the cell type transition matrix. We then transposed this matrix, as PhyloVelo reverses directionality in its transitions, and row normalized it as well.
For the TLS dataset, we used an AnnData object generated by a standard Seurat reciprocal principal component analysis (rPCA) integration pipeline of the scRNA-seq data for the sequenced TLS experiments29. This pipeline normalizes counts for 22,291 genes and generates uniform manifold approximation and projection (UMAP) coordinates for each cell. We subsetted the AnnData object to cells that are in the cell lineage trees and calculated the depth of each cell as the number of edges from the root that have at least one mutation. We further removed genes with a count lower than 50 across all cells. We used the Scanpy package in Python for the manipulation of the AnnData object and the thresholding of genes.
For the dataset from Weinreb et al., we generated an AnnData object using the normalized gene counts from the publicly available data in the original study30. We included only cells with barcodes. We then closely followed the analysis suggested in the documentation of ref. 54 (https://phylovelo.readthedocs.io/en/latest/notebook/in_vitro_hematopoiesis.html), using largely the same parameter choices. One notable difference is that we used n_neigh = 500 in the ‘velocity_embedding’ function, as using the originally specified 100 generates an error in state graph construction.
Choosing Carta optimal number of progenitors in real data
We selected the number k* of progenitors by finding an elbow in the k versus minimum discrepancy graph, using the kneedle algorithm. Initially, the kneedle algorithm found elbow points with very few progenitors (k = 4 for the DAG curve for the TLS data and k = 3 for the tree curve for the data from Weinreb et al., respectively). These elbows provided cell differentiation maps that included too few progenitors to fully capture the complex dynamics in the developmental systems that we explored. We found kneedle to be conservative, selecting an elbow at the first point with a substantial reduction in the difference in discrepancy with the previous point. Hence, we sought to select an elbow among the ‘flat’ region of each curve to determine which progenitors whose inclusion yields the lowest value in terms of reduced discrepancy while maintaining a useful number of progenitors. Thus, we applied kneedle to the regions where the curve flattens out (k = 4, …11 for the DAG curve for the TLS data and k = 5, …11 for the tree curve for the data from Weinreb et al.), giving elbows at k = 7 for both datasets.
Choosing edges in Carta real data cell differentiation maps
For the Carta-DAG cell differentiation map with k = 7 inferred for TLS (Fig. 3d), we include all transitions that appear frequently in the cell lineage trees. Specifically, we define the ‘cellular flow’ \(w(P,{P}^{{\prime} })\) for a transition \((P,{P}^{{\prime} })\) as the number of cells across the given set of cell lineage trees that traverse through that transition. To calculate the cellular flow for a transition, we counted the instances in which \(\ell (v)=P,\ell (u)={P}^{{\prime} }\) for each edge (u, v) in a cell lineage tree T, weighting by the number of leaf descendants of v. This weighting preserves flow in the map such that the cellular flow entering a progenitor is equal to the cellular flow exiting it—that is, \({\sum }_{({P}^{{\prime} },P)\in {F}_{S}}w({P}^{{\prime} },P)={\sum }_{(P,{P}^{{\prime\prime} })\in {F}_{S}}w(P,{P}^{{\prime\prime} })\). We keep an edge in the cell differentiation map in Fig. 3d if (1) the edge is necessary to ensure that an extant state is reachable by a progenitor that includes that in its potency or (2) the edge has a cellular flow that is >0.2 × deg+(P), meaning that the edge accounts for more than 20% of the cellular flow from its parent progenitor. Note that this criterion also removes 0-flow edges. For the Carta-Tree cell differentiation map with k = 7 inferred for the data from Weinreb et al., we only include edges such that the map has a tree structure.
Discrepancy of existing method-inferred differentiation maps
The first step in calculating the discrepancy of cell differentiation maps inferred by existing methods is determining the potencies of progenitors in the inferred cell differentiation maps. For methods that produce binary-tree-structured cell differentiation maps (ICE-FASE and EvoC), the potency of a progenitor—that is, an internal vertex v of the map—is the set of observed cell types—that is, leaves—in the subtree rooted at v. As Fitch and PhyloVelo do not explicitly infer progenitors, we devise a scheme to obtain progenitors from their cell differentiation maps. The output of Fitch and PhyloVelo is a normalized transition frequency (f(ti, tj)) between each pair of states ti, tj ∈ S. For each observed cell type ti, we introduce a progenitor as {tj: f(ti, tj) ≥ ϵ}. This is the set of each cell type j for which the transition frequency from a cell type i exceeds threshold ϵ. In this work, we chose \(\epsilon =\frac{1}{| S| }\), and, thus, ϵ = 0.166 for the TLS data and ϵ = 0.111 for the data from Weinreb et al.
We computed the discrepancy for each method by solving the PLP (Supplementary Section 1) using the dynamic programming algorithm. The number of cell type transitions (Fig. 3f,g) is determined by the inferred minimum discrepancy labeling of the cell lineage trees.
Calculating undifferentiated cell distances in Weinreb et al. data
We labeled each undifferentiated cell (cells labeled with the ‘undifferentiated’ cell type in ref. 30) in the data from Weinreb et al. with the progenitor type that Carta assigns to the ancestral cell of its star-shaped cell lineage tree; these are the labels shown in Fig. 5a. We next describe how we calculate the distance of each undifferentiated cell to the closest observed cell type cluster in gene expression space. First, we obtained the normalized counts for 25,289 genes across all cells in this dataset from the publicly available in vitro differentiation timecourse data from Weinreb et al.30 (https://github.com/AllonKleinLab/paper-data/tree/master/Lineage_tracing_on_transcriptional_landscapes_links_state_to_fate_during_differentiation). We then removed cells with counts = 0 or counts > 1,000,000 and performed PCA with n = 50 components. We next calculated a 50-principal-component centroid for each observed cell type by averaging across the principal component values of cells of that type and then calculated the Euclidean distance in principal component values between each undifferentiated cell and each centroid. Finally, in Fig. 5c, for each observed cell type cluster, we calculated the proportion of undifferentiated cells labeled with each progenitor cell type by Carta that is closest to that cluster.
Statistics and reproducibility
The TLS dataset generated by Bolondi et al.29 includes 14 single cell lineages with a total of 4,709 extant cells across all lineages. The dataset of mouse HSCs generated by Weinreb et al. includes 5,864 uniquely barcoded clones (star-shaped cell lineage trees) with a total of 49,302 cells. No statistical methods were used to predetermine the sample size of these datasets. The experimental data were obtained from other sources, and we did not attempt to experimentally replicate the data in our study. For the results of our computational experiments, we perform robustness analyses that show the result of our method on various subsamplings and definitions of cell types for both the TLS dataset as well as the mouse hematopoeisis dataset (Supplementary Sections 8–10). Samples were not allocated into different groups for statistical testing, and, hence, randomization and blinding were not used in the experimental data collection process. In the TLS dataset, we did not exclude any data. In the mouse hematopoietic dataset, we observed 107 different unique combinations of cell types descending from one root (observed potency of that root). This was a larger number than expected given that the data have only nine extant cell types. We reasoned that there would be many spurious observed potencies that are the result of two factors. First, these barcoded clones taken as cell lineage trees are small (averaging 8.4 cells per lineage tree), and, hence, it is likely that some of these observed potencies may be subsets of larger potencies that are not observed due to low sampling. Second, these cell lineage trees are shallow (depth 1), and, hence, some of these observed potencies may be supersets of progenitors with smaller and more restricted potencies that would have been resolved by trees with higher depth and resolution. Hence, we performed a mild filtering and removed all observed potencies in this dataset that had low data support (appeared in fewer than 10 clones). This process removed 4.1% of clones.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Simulated data are available on GitHub at https://github.com/raphael-group/CARTA/tree/main/data/simulations. Raw sequencing data for the TLS dataset are available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE220949. The processed lineage trees and cell type annotations are available at https://github.com/raphael-group/CARTA/tree/main/data/TLS. The single-cell lineage tracing data of mouse hematopoiesis are available at https://github.com/AllonKleinLab/paper-data/tree/master/Lineage_tracing_on_transcriptional_landscapes_links_state_to_fate_during_differentiation.
Code availability
The code is publicly available at https://github.com/raphael-group/CARTA under a BSD 3-Clause license.
References
Kimmel, C. B., Warga, R. M. & Schilling, T. F. Origin and organization of the zebrafish fate map. Development 108, 581–594 (1990).
Axelrod, D. Carbocyanine dye orientation in red cell membrane studied by microscopic fluorescence polarization. Biophys. J. 26, 557–573 (1979).
Cotsarelis, G., Sun, T.-T. & Lavker, R. M. Label-retaining cells reside in the bulge area of pilosebaceous unit: implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell 61, 1329–1337 (1990).
Tumbar, T. et al. Defining the epithelial stem cell niche in skin. Science 303, 359–363 (2004).
Kretzschmar, K. & Watt, F. M. Lineage tracing. Cell 148, 33–45 (2012).
Sulston, J. E., Schierenberg, E., White, J. G. & Thomson, J. N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64–119 (1983).
Forrow, A. & Schiebinger, G. LineageOT is a unified framework for lineage tracing and trajectory inference. Nat. Commun. 12, 4940 (2021).
Schiebinger, G. et al. Optimal-transport analysis of single-cell gene expression identifies developmental trajectories in reprogramming. Cell 176, 928–943 (2019).
Pellin, D. et al. A comprehensive single cell transcriptional landscape of human hematopoietic progenitors. Nat. Commun. 10, 2395 (2019).
Tusi, B. K. et al. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature 555, 54–60 (2018).
Buenrostro, J. D. et al. Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell 173, 1535–1548 (2018).
Qiu, C. et al. A single-cell time-lapse of mouse prenatal development from gastrula to birth. Nature 626, 1084–1093 (2024).
Bendall, S. C. et al. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157, 714–725 (2014).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017).
Wolf, F. A. et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59 (2019).
Welch, J. D., Hartemink, A. J. & Prins, J. F. SLICER: inferring branched, nonlinear cellular trajectories from single cell RNA-seq data. Genome Biol. 17, 106 (2016).
Street, K. et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018).
Chen, H. et al. Single-cell trajectories reconstruction, exploration and mapping of omics data with STREAM. Nat. Commun. 10, 1903 (2019).
Herman, J. S., Sagar, n & Gruen, D. FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nat. Methods 15, 379–386 (2018).
Setty, M. et al. Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat. Biotechnol. 34, 637–645 (2016).
Saelens, W., Cannoodt, R., Todorov, H. & Saeys, Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 37, 547–554 (2019).
Wagner, D. E. & Klein, A. M. Lineage tracing meets single-cell omics: opportunities and challenges. Nat. Rev. Genet. 21, 410–427 (2020).
Wang, L. et al. Current progress and potential opportunities to infer single-cell developmental trajectory and cell fate. Curr. Opin. Syst. Biol. 26, 1–11 (2021).
Deconinck, L., Cannoodt, R., Saelens, W., Deplancke, B. & Saeys, Y. Recent advances in trajectory inference from single-cell omics data. Curr. Opin. Syst. Biol. 27, 100344 (2021).
Wagner, D. E. et al. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science 360, 981–987 (2018).
Veling, M. W. et al. Identification of neuronal lineages in the Drosophila peripheral nervous system with a ‘digital’ multi-spectral lineage tracing system. Cell Rep. 29, 3303–3312 (2019).
Chan, M. M. et al. Molecular recording of mammalian embryogenesis. Nature 570, 77–82 (2019).
Bolondi, A. et al. Reconstructing axial progenitor field dynamics in mouse stem cell-derived embryoids. Dev. Cell 59, 1489–1505 (2024).
Weinreb, C., Rodriguez-Fraticelli, A., Camargo, F. D. & Klein, A. M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 367, eaaw3381 (2020).
He, Z. et al. Lineage recording in human cerebral organoids. Nat. Methods 19, 90–99 (2022).
Kong, W. et al. CellTagging: combinatorial indexing to simultaneously map lineage and identity at single-cell resolution. Nat. Protoc. 15, 750–772 (2020).
Jindal, K. et al. Single-cell lineage capture across genomic modalities with CellTag-multi reveals fate-specific gene regulatory changes. Nat. Biotechnol. 42, 946–959 (2023).
McKenna, A. et al. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353, aaf7907 (2016).
Raj, B., Gagnon, J. A. & Schier, A. F. Large-scale reconstruction of cell lineages using single-cell readout of transcriptomes and CRISPR–Cas9 barcodes by scGESTALT. Nat. Protoc. 13, 2685–2713 (2018).
Alemany, A., Florescu, M., Baron, C. S., Peterson-Maduro, J. & Van Oudenaarden, A. Whole-organism clone tracing using single-cell sequencing. Nature 556, 108–112 (2018).
Spanjaard, B. et al. Simultaneous lineage tracing and cell-type identification using CRISPR–Cas9-induced genetic scars. Nat. Biotechnol. 36, 469–473 (2018).
Quinn, J. J. et al. Single-cell lineages reveal the rates, routes, and drivers of metastasis in cancer xenografts. Science 371, eabc1944 (2021).
Yang, D. et al. Lineage tracing reveals the phylodynamics, plasticity, and paths of tumor evolution. Cell 185, 1905–1923 (2022).
Jones, M. G. et al. Inference of single-cell phylogenies from lineage tracing data using Cassiopeia. Genome Biol. 21, 92 (2020).
Sashittal, P., Schmidt, H., Chan, M. & Raphael, B. J. Startle: a star homoplasy approach for CRISPR-Cas9 lineage tracing. Cell Syst. 14, 1113–1121 (2023).
Pan, X., Li, H., Putta, P. & Zhang, X. LinRace: cell division history reconstruction of single cells using paired lineage barcode and gene expression data. Nat. Commun. 14, 8388 (2023).
Feng, J. et al. Estimation of cell lineage trees by maximum-likelihood phylogenetics. Ann. Appl. Stat. 15, 343 (2021).
Gong, W. et al. Benchmarked approaches for reconstruction of in vitro cell lineages and in silico models of C. elegans and M. musculus developmental trees. Cell Syst. 12, 810–826 (2021).
Zafar, H., Lin, C. & Bar-Joseph, Z. Single-cell lineage tracing by integrating CRISPR–Cas9 mutations with transcriptomic data. Nat. Commun. 11, 3055 (2020).
Chu, G., Mai, U., Schmidt, H. & Raphael, B. J. Maximum likelihood inference of time-scaled cell lineage trees with mixed-type missing data using LAML. Genome Biol. 26, 189 (2025).
Fang, W. et al. Quantitative fate mapping: a general framework for analyzing progenitor state dynamics via retrospective lineage barcoding. Cell 185, 4604–4620 (2022).
Veenvliet, J. V. et al. Mouse embryonic stem cells self-organize into trunk-like structures with neural tube and somites. Science 370, eaba4937 (2020).
Bolondi, A. et al. Generation of mouse pluripotent stem cell-derived trunk-like structures: an in vitro model of post-implantation embryogenesis. Bio Protoc. 11, e4042 (2021).
Feller, W. Die Grundlagen der Volterraschen Theorie des Kampfes ums Dasein in wahrscheinlichkeitstheoretischer Behandlung. Acta Biotheoretica 5, 11–40 (1939).
Bolkent, S. Cellular and molecular mechanisms of asymmetric stem cell division in tissue homeostasis. Genes Cells 29, 1099–1110 (2024).
Sunchu, B. & Cabernard, C. Principles and mechanisms of asymmetric cell division. Development 147, dev167650 (2020).
Jaccard, P. The distribution of the flora in the alpine zone. 1. New Phytologist 11, 37–50 (1912).
Wang, K. et al. PhyloVelo enhances transcriptomic velocity field mapping using monotonically expressed genes. Nat. Biotechnol. 42, 778–789 (2023).
Lawson, K. A., Meneses, J. J. & Pedersen, R. A. Clonal analysis of epiblast fate during germ layer formation in the mouse embryo. Development 113, 891–911 (1991).
Forlani, S., Lawson, K. A. & Deschamps, J. Acquisition of Hox codes during gastrulation and axial elongation in the mouse embryo. Development 130, 3807–3819 (2003).
Solovieva, T., Wilson, V. & Stern, C. D. A niche for axial stem cells—a cellular perspective in amniotes. Dev. Biol. 490, 13–21 (2022).
Lagha, M. et al. Pax3:Foxc2 reciprocal repression in the somite modulates muscular versus vascular cell fate choice in multipotent progenitors. Dev. Cell 17, 892–899 (2009).
Nguyen, P. D. et al. Haematopoietic stem cell induction by somite-derived endothelial cells controlled by meox1. Nature 512, 314–318 (2014).
Villani, A.-C. et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, eaah4573 (2017).
Balan, S., Saxena, M. & Bhardwaj, N. Dendritic cell subsets and locations. Int. Rev. Cell Mol. Biol. 348, 1–68 (2019).
Chen, B., Zhu, L., Yang, S. & Su, W. Unraveling the heterogeneity and ontogeny of dendritic cells using single-cell RNA sequencing. Front. Immunol. 12, 711329 (2021).
Fitch, W. M. Toward defining the course of evolution: minimum change for a specific tree topology. Syst. Biol. 20, 406–416 (1971).
Seita, J. & Weissman, I. L. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2, 640–653 (2010).
Helft, J. et al. Dendritic cell lineage potential in human early hematopoietic progenitors. Cell Rep. 20, 529–537 (2017).
Olson, O. C., Kang, Y.-A. & Passegué, E. Normal hematopoiesis is a balancing act of self-renewal and regeneration. Cold Spring Harb. Perspect. Med. 10, a035519 (2020).
Karamitros, D. et al. Single-cell analysis reveals the continuum of human lympho-myeloid progenitor cells. Nat. Immunol. 19, 85–97 (2018).
Dong, F. et al. Differentiation of transplanted haematopoietic stem cells tracked by single-cell transcriptomic analysis. Nat. Cell Biol. 22, 630–639 (2020).
Metcalf, D. Hematopoietic cytokines. Blood 111, 485–491 (2008).
Zaro, B. W. et al. Proteomic analysis of young and old mouse hematopoietic stem cells and their progenitors reveals post-transcriptional regulation in stem cells. eLife 9, e62210 (2020).
Cheng, H., Zheng, Z. & Cheng, T. New paradigms on hematopoietic stem cell differentiation. Protein Cell 11, 34–44 (2020).
Carrelha, J. et al. Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature 554, 106–111 (2018).
Yamamoto, R. et al. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 154, 1112–1126 (2013).
Robinson, D. F. & Foulds, L. R. Comparison of phylogenetic trees. Math. Biosci. 53, 131–147 (1981).
Koller, D. & Friedman, N. Probabilistic Graphical Models: Principles and Techniques (MIT Press, 2009).
Li, J. & Stanger, B. Z. How tumor cell dedifferentiation drives immune evasion and resistance to immunotherapy. Cancer Res. 80, 4037–4041 (2020).
Sell, S. Cellular origin of cancer: dedifferentiation or stem cell maturation arrest? Environ. Health Perspect. 101, 15–26 (1993).
Yamada, Y., Haga, H. & Yamada, Y. Concise review: dedifferentiation meets cancer development: proof of concept for epigenetic cancer. Stem Cells Transl. Med. 3, 1182–1187 (2014).
Friedmann-Morvinski, D. & Verma, I. M. Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO Rep. 15, 244–253 (2014).
Chow, K.-H. K. et al. Imaging cell lineage with a synthetic digital recording system. Science 372, eabb3099 (2021).
Chadly, D. M. et al. Reconstructing cell histories in space with image-readable base editor recording. Preprint at bioRxiv https://doi.org/10.1101/2024.01.03.573434 (2024).
Li, L. et al. A mouse model with high clonal barcode diversity for joint lineage, transcriptomic, and epigenomic profiling in single cells. Cell 186, 5183–5199 (2023).
Weinreb, C., Wolock, S. & Klein, A. M. SPRING: a kinetic interface for visualizing high dimensional single-cell expression data. Bioinformatics 34, 1246–1248 (2018).
Sankoff, D. & Rousseau, P. Locating the vertices of a steiner tree in an arbitrary metric space. Mathematical Programming 9, 240–246 (1975).
Satopaa, V., Albrecht, J., Irwin, D. & Raghavan, B. Finding a ‘kneedle’ in a haystack: detecting knee points in system behavior. In 2011 31st International Conference on Distributed Computing Systems Workshops 166–171 https://doi.org/10.1109/ICDCSW.2011.20 (IEEE, 2011).
Gurobi Optimization. Gurobi Optimizer Reference Manual. https://docs.gurobi.com/projects/optimizer/en/current/index.html
Prillo, S., Ravoor, A., Yosef, N., & Song, Y. S. (2025). ConvexML: Fast and accurate branch length estimation under irreversible mutation models, illustrated through applications to CRISPR/Cas9-based lineage tracing. Syst. Biol. syaf054 (2025).
Robinson, D. J. An Introduction to Abstract Algebra (Walter de Gruyter, 2003).
Sokal, R. R. & Michener, C. D. A statistical method for evaluating systematic relationships. Kans. Univ. Sci. Bull. 38, 1409–1438 (1958).
Qiu, X. et al. Mapping transcriptomic vector fields of single cells. Cell 185, 690–711 (2022).
Acknowledgements
This work is supported by grant 2024-345885 from the Chan Zuckerberg Initiative DAF, an advised fund of the Silicon Valley Community Foundation; National Institutes of Health grant DP2HD111537 to M.M.C.; National Cancer Institute grant U24CA248453 to B.J.R.; and the Princeton Catalysis Initiative. R.Z. is supported by National Human Genome Research Institute training grant T32HG003284.
Author information
Authors and Affiliations
Contributions
P.S., R.Z., M.M.C. and B.J.R. conceived and developed the method. P.S., R.Z. and H.S. implemented the software. P.S. and R.Z. performed analysis of the lineage tracing datasets. B.L. produced the cell lineage trees and cell type annotations for one of the lineage tracing datasets (TLS). A.S. ran competing methods on lineage tracing datasets. P.S. and H.S. derived the complexity proofs. P.S., R.Z., A.B., M.M.C. and B.J.R. interpreted the results and wrote the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks Wouter Saelens and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Madhura Mukhopadhyay, in collaboration with the Nature Methods team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Graphical representations of cell differentiation maps inferred by Carta on the TLS data.
(a) k = 5, (b) k = 6, (c) k = 7 and (d) k = 8 progenitors. Black: edges in the transitive reduction of the graph. Red: edges not in the transitive reduction of the graph.
Extended Data Fig. 2 Graphical representations of cell differentiation maps inferred for the TLS dataset by other methods.
(a) Carta-Tree cell differentiation map. (b) ICE-FASE cell differentiation map. (c) EvoC cell differentiation map. (d) Fitch cell differentiation map. Edge weights indicate the normalized transition frequency from one observed cell type to another. Only edges with frequency ≥1/∣S∣ = 0.166 are shown.
Extended Data Fig. 3 Graphical representations of cell differentiation maps inferred by Carta on the mouse hematopoiesis data from Weinreb et al.
(a) k = 5, (b) k = 6, (c) k = 7 and (d) k = 8 progenitors. Red indicates inferred progenitors that are non-canonical.
Extended Data Fig. 4 Graphical representations of cell differentiation maps inferred for the mouse hematopoiesis data from Weinreb et al. by other methods.
(a) ICE-FASE cell differentiation map. Red indicates inferred progenitors that are non-canonical, that is do not agree with the canonical model. (b) EvoC cell differentiation map. Red indicates inferred progenitors that are non-canonical, that is do not agree with the canonical model. (c) PhyloVelo cell differentiation map. Edge weights indicate the normalized transition frequency from one observed cell type to another. Only edges with frequency ≥1/∣S∣ = 0.111 are shown. (d) Fitch cell differentiation map. Edge weights indicate the normalized transition frequency from one observed cell type to another. Only edges with frequency ≥1/∣S∣ = 0.111 are shown.
Extended Data Fig. 5 Progenitor predictions given by Carta (colored dots) for undifferentiated cells sampled at different days.
(a) Day 2 (358 cells), (b) day 4 (3561 cells) and (c) day 6 (7651 cells).
Supplementary information
Supplementary Information (download PDF )
Supplementary Sections 1–11, Supplementary Tables 1 and 2 and Supplementary Figs. 1–11.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sashittal, P., Zhang, R.Y., Law, B.K. et al. Inferring cell differentiation maps from lineage tracing data. Nat Methods 23, 532–541 (2026). https://doi.org/10.1038/s41592-025-02903-z
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41592-025-02903-z
