
Abstract
Designing new enzymes typically begins with idealized arrangements of catalytic functional groups around a reaction transition state, then attempts to generate protein structures that precisely position these groups. Current AI-based methods can create active enzymes but require predefined residue positions and rely on reverse-building residue backbones from side-chain placements, which limits design flexibility. Here we show that a new deep generative model, RoseTTAFold diffusion 2 (RFdiffusion2), overcomes these constraints by designing enzymes directly from functional group geometries without specifying residue order or performing inverse rotamer generation. RFdiffusion2 successfully generates scaffolds for all 41 active sites in a diverse benchmark, compared to 16 using previous methods. We further design enzymes for three distinct catalytic mechanisms and identify active candidates after experimentally testing fewer than 96 sequences in each case. These results highlight the potential of atomic-level generative modeling to create de novo enzymes directly from reaction mechanisms.
Similar content being viewed by others

Main
A grand challenge in de novo protein design is the generation of enzymes that catalyze novel reactions. De novo enzyme design starts from a detailed description of an active site composition and geometry predicted to catalyze the reaction of interest. This active site description, called a theozyme, describes placement of protein functional groups around the reaction transition state(s) and any reaction cofactors1,2. The de novo enzyme design task is to generate protein scaffolds that accommodate such theozymes. Pre-deep learning methods such as RosettaMatch searched through sets of already existing native or designed3 scaffolds for possible placements of the catalytic residues. While many enzymes were designed using this approach, it was restricted to theozyme geometries that could be matched to the input scaffold set4. Advances in deep learning with diffusion models5,6,7,8 have removed the need for scaffold libraries for many substructure scaffolding tasks by directly sampling diverse proteins containing the desired substructure (motif) through a technique known as motif scaffolding9,10,11,12. However, thus far, these methods all operate on a backbone-level representation of proteins as a series of amino acid residues and, consequently, can only scaffold motifs represented at the backbone level.
Current approaches attempt to overcome this limitation for atom-level active site descriptions by enumerating possible conformations and sequence indices for the catalytic residues and then in a separate step using motif scaffolding to generate proteins that scaffold these backbone positions13,14. While active enzymes have been generated using this approach, it is computationally inefficient and does not scale to more complex active sites as the number of combinations of backbone coordinates to scaffold grows exponentially with the number of catalytic residues. A method capable of scaffolding complex theozymes described at the atom level would have widespread applications for enzyme design and beyond15,16,17.
We reasoned that substantially improved performance on more complex enzyme active site scaffolding challenges could be achieved by a generative model capable of selecting the conformations and sequence indices of the catalytic residues by modeling the full joint distribution of rotamers, sequence indices and scaffolds conditioned directly on atom-level active site descriptions. With RFdiffusion2, we set out to extend RoseTTAFold diffusion All-Atom (RFdiffusionAA)18 to generate structures conditioned on these minimal active site descriptions (Fig. 1).

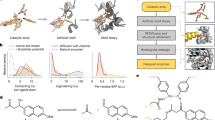
a, De novo enzyme design starts from a configuration of catalytic groups around the reaction transition state(s) (a theozyme) generated using quantum chemistry, protein structural analysis and/or chemical reasoning. b, RFdiffusion2 generates protein structures that support the theozyme. In row 1, the backbone trajectory shows the amino acid residue frames (pastel) as they transform from a sample drawn from the noise distribution into a protein backbone. Row 2—a zoom-in of row 1—shows the non-motif side-chain atoms (slate gray) connecting the atomic motif (teal) with the protein backbone. At t = 1 the intra-residue bonds are shown for the atomized residues. Right, The distances between the Cα coordinates of the unindexed, atomized residues and the backbone residues they superimpose at t = 1. Over the course of the trajectory, the model matches these unindexed residues to indexed residues of the protein backbone, such that by the end of the trajectory the unindexed residue’s Cα occupies the same location as the Cα of the protein backbone in Euclidean space. c, The design pipeline starts from the input theozyme, followed by RFdiffusion2 to generate the structure, and LigandMPNN to generate amino acid sequences that encode the structure and stabilize the transition state. Designs are evaluated by all-atom structure prediction (for example, using Chai-1 and AF3) and are considered an in silico success if the design (pastel) and prediction (light gray) align to a sufficient degree. Two representative examples of consistency between design model and predicted structure at the level we take to constitute a success are shown in the right panels. The two cases pictured are the creatinase and taurine dioxygenase motifs from the AME benchmark described in ‘AME benchmark’ in the Results (AME IDs: M0096_1chm AND M0129_1os7).
Atomic motif conditioning
To address this challenge, we sought to generalize motif scaffolding beyond sequence indexed, backbone-level motifs. Prior motif-scaffolding methods represent motifs as ‘backbone frames’, in which each amino acid’s N, Cα, C backbone atoms are parameterized as an element in SE(3). Each motif frame requires a prespecified sequence index that indicates the frame’s location along the backbone chain. In contrast, the protein component of an atom-level active site description includes only the side-chain functional group atoms, not the backbone, of the participating amino acid residues; these residues can be anywhere along the sequence and hence do not have a specified index. While the indexed frame representation is sufficient for scaffolding large contiguous domains that can be accurately described at the residue level, it is insufficient to express the task of scaffolding disconnected groups of atoms belonging to residues of unknown indices.
Our approach is to create an extended representation with differing levels of resolution and index information that is capable of expressing more complex motifs. In RFdiffusion2, we use the RoseTTAFold All-Atom neural network architecture in which each residue in the input and output can be represented as a frame or as heavy atom coordinates (an atomized residue)18. During training, we represent some residues with frames and atomize others. For each atomized residue, the network learns to model the distribution of side-chain poses. By providing known coordinates for some side-chain atoms, which we term the atomic motif, the network learns to model the distribution of proteins conditioned on the inclusion of such atomic substructures. At inference time, we can then condition on the coordinates of individual protein atoms such as the side-chain (or backbone) functional groups present in a theozyme, along with any ligand atoms representing transition state geometry. This ability to condition on individual atoms rather than entire residues allows us to forgo inverse rotamer sampling4 used in previous approaches and instead allow the model to simultaneously infer an appropriate rotamer and scaffold.
We can further extend the representation to remove the need to know the sequence indices of a motif to scaffold it. During training, we select subsets of residues, duplicate them and remove their index features to create ‘unindexed residues’. Without any auxiliary losses, the network learns that unindexed residues are always superimposed on indexed residues in unnoised structures. By providing coordinates for unindexed residues, which we term an ‘unindexed motif’, the network learns to model the distribution of proteins conditioned on the inclusion of a known substructure at unknown indices. At inference time, we then have the flexibility to condition on a motif consisting of residues with known or unknown indices because theozymes do not prescribe the sequence indices of their constituent residues. The ability to condition on motifs without specifying their indices enables us to forgo naive index sampling used in previous approaches and instead allow the model to simultaneously infer indices for the motif while scaffolding it (Fig. 2b).

a, In the original RFdiffusion, two preprocessing steps are required to transform an unindexed atomic motif into a suitable input. These steps—inverse rotamer sampling and sequence index sampling—both require selecting from an exponentially large search spaces of L!/(L − M)! and M(no. of possible rotamer states), respectively, where L is the number of residues, while M is the number of residues that are needed for the active site. RFdiffusion2 does not require such preprocessing steps and can scaffold unindexed atomic motifs directly. b, RFdiffusion2 can be conditioned on motifs in different representations. Three versions of the same motif (M0904; PDB 1QGX) from AME are shown on the leftmost column, different backbone samples in the middle columns, and the resulting diversity of sequence indices and rotamers on the rightmost columns. (i) The backbone motif includes a prespecified rotamer and index as required by RFdiffusion. (ii) The atomic motif has prespecified sequence indices but unspecified side-chain conformation. (iii) Only unindexed atom positions are provided, not the residue indices or side-chain rotamer conformations. The rotamer and sequence indices are sampled during the RFdiffusion2 trajectories, increasing the diversity of possible solutions to the motif-scaffolding problem. c, Each ligand atom can be labeled with a RASA category to control how solvent exposed the ligand is. The example RASA conditions are in the left column, a backbone sample with the ligand in the middle, and the distribution of ligand atom RASA from 100 designs with the RASA condition. When all atoms are labeled as exposed, the ligand RASA is concentrated around 1.0 and the backbone does not come into contact with the ligand. Conversely, when all atoms are labeled as buried, the ligand RASA is concentrated around 0; the sample shows the backbone almost completely covering the ligand. Labeling half the ligand as exposed and the other half as buried leads to RFdiffusion2 generating backbones that only bind to the buried side of the ligand. d, RFdiffusion2 can be provided with an ORI token that specifies the desired center of mass (CoM) of the scaffold with respect to the ligand. Two different ORI positions are shown in the left column. The middle column shows samples with scaffolds centered at the indicated ORI token positions. The distribution of CoMs from 100 sampled designs with the ORI token show that the scaffolds generally follow the ORI condition of where to place the scaffold. e, RFdiffusion2 can be provided with partial ligand input in which case it must sample the remaining ligand degrees of freedom while generating the protein. The left column shows the partial ligand input. The middle column shows in gray a conformer along with the protein generated by RFdiffusion2. Finally, the right column shows the distribution of ten generated conformers. In Supplementary Fig. 3, we analyze the physical plausibility of the generated conformers.
The resulting model, RFdiffusion2, can generate proteins conditioned on a broad range of motifs including side-chain motifs, motifs without known sequence indices and ligand motifs. When training RFdiffusion and RFDiffusionAA, we found performance started to worsen over extended periods of training (Supplementary Fig. 1). Through a combination of auxiliary losses and self-conditioning19, these methods were able to achieve high success rates on their respective tasks in short training sessions. Due to the complexity of the unindexed atomic motif-scaffolding task, we expected that a stable objective would be required. To this end, we train RFdiffusion2 with flow matching7,8, a simpler framework for diffusion models20,21 shown empirically to have improved training and generation efficiency in other domains22. Briefly, this framework interpolates a training example toward a noise sample and trains a neural network to denoise by predicting the original, uncorrupted example. If trained to denoise with sufficient accuracy, the model can sample from the data distribution by iteratively denoising a sample drawn from the noise prior. Our representation of the data distribution contains both atoms and backbone frames that are elements of \({{\mathbb{R}}}^{3}\) and SE(3), respectively. Flow matching on \({{\mathbb{R}}}^{3}\) follows its original derivation using Gaussian probability paths, while for SE(3) we follow the formulation in FrameFlow23,24 that utilizes Riemannian flow matching25 and removes approximations for rotational losses present in the RFdiffusion9. With these improvements, RFdiffusion2 trained stably from randomly initialized neural network weights, and does not require auxiliary losses or use self-conditioning. Decoupling RFdiffusion2 from structure prediction removes constraints around the neural network architecture and enables new generative modeling tasks.
Our training dataset consists of biomolecular structures from the Protein Data Bank (PDB)26 that include proteins, protein–small-molecule complexes, protein–metal complexes and covalently modified proteins, filtering out common solvents and crystallization additives18. Each structure undergoes a motif extraction procedure to construct a motif-scaffolding training example. First, we select a random subset of residues to be the motif. Motif residues are chosen uniformly at random to ensure RFdiffusion2 sees a diverse range of interatomic geometries in the motif. Second, each motif residue is represented as either a frame or an atomized residue. If a motif residue is atomized, only a random subgraph of the amino acid heavy atoms are provided as the motif. Third, we decide whether the motif will be featurized as indexed or unindexed. Lastly, for any ligand present, we sample a random connected subgraph of its heavy atoms to be provided as a motif with relative accessible surface area (RASA)27,28 labels for each atom in the ligand intermittently provided. We resample the motif on each training step as a form of data augmentation to ensure that the same noised structure is not always shown alongside the same motif. We train the final model for 17 days using 24 A100 Nvidia GPUs.
In our early experiments with flow matching, we found that the choice of how the ground-truth structures were centered relative to the origin in training substantially affected the quality of inference outputs. A natural strategy is to globally center each ground-truth structure; however, this leaks the offset between the scaffold center of mass and the motif. At inference time, this strategy requires the exact specification of the desired offset, which is usually not known. A common fix for this pathology in motif-scaffolding diffusion models is to center ground-truth structures on the motif, allowing the model to determine the offset between the scaffold center of mass and the motif12,24. However even when the motif is centered, due to the interpolation scheme used in flow matching, the model is able to exactly determine this offset from any partially noised structure. The resultant behavior is that in the first denoising step, in which the network receives pure noise, it predicts an offset that it does not learn to refine in subsequent denoising steps. We instead introduce ‘stochastic centering’, which first centers the ground-truth structure and adds a small global translation sampled from a three-dimensional Gaussian such that the noised input structures only encode an approximate offset between the motif and scaffold. This enables the model to refine the placement of the motif within the overall structure over the course of the inference trajectory. At inference time, users supply a prior belief about the placement of the motif through a special ORI pseudo-atom that specifies the approximate center of mass of the generated structure. This enables enzyme designers to control the active site and transition state orientation relative to the protein core (Fig. 2d). For example, given an elongated small-molecule or transition state with one end quite polar or charged, placement of the ORI token adjacent to the opposite end (or displaced from this end along a vector running through the long axis of the molecule) results in a designed binder or enzyme with a binding pocket extending radially from the center of the protein with the polar/charged end of the small molecule exposed to solvent.
RFdiffusion2 provides two additional conditioning capabilities of the ligand that are useful in de novo enzyme design. First, to provide finer control over the depth at which each reactant and/or cofactor is buried within the protein, we enable users to specify the RASA of each atom. By providing the RASA of each ligand atom 50% of the time during training, RFdiffusion2 learns to generate structures that respect those atom-wise conditions at inference time when provided (Fig. 2c). Second, the user may know the ligand atoms of a transition state but may not know the full ligand conformer. We allow the user to specify ‘partial ligands’ where only the known ligand atoms are provided while RFdiffusion2 infers the rest of the ligand conformer. Analysis of the physical plausibility of the generated conformers shows they match closely with RDkit-generated conformers (Supplementary Fig. 3). We find RFdiffusion2 can, given partial ligand coordinates, sample physically realistic conformers for the entire ligand (Fig. 2d), removing the need to prespecify the complete ligand conformer with an external tool. Together, the conditioning capabilities open up greater control over the geometric properties of the protein–ligand complex during inference.
Results
AME benchmark
An in silico enzyme motif-scaffolding benchmark was introduced in RFdiffusion9,29 which contained one active site from each of the five most represented Enzyme Commission (EC) classes in the Mechanism and Catalytic Site Atlas (M-CSA)30. We find that this benchmark does not accurately reflect the challenges of de novo enzyme design due to its use of indexed backbone motifs, lack of ligands and lack of active site diversity with only five enzyme cases. To evaluate RFdiffusion2, we developed a new benchmark that better reflects the theozyme scaffolding problem of de novo enzyme design.
We cross-referenced the 958 hand-curated catalytic active sites downloaded from the M-CSA with the Proportion of Atoms Residing In Identical Topology (PARITY)31 dataset and selected reactions with PDB crystal structures where all reactants and cofactors were present. After curation, we found 41 active sites spanning a diverse set of reactions in EC29 classes 1–5 (Fig. 3a; EC classes 1–5 account for 96% of examples in M-CSA). The annotations in M-CSA are at the residue level, so we extracted random connected sets of atoms from each catalytic residue to treat as the protein component of the theozyme for each benchmark case (Fig. 3b).

a, EC number distribution of the AME benchmark30. b, In silico success rate of RFdiffusion2 and RFdiffusion as a function of the discontiguous chain segments containing motif atoms (the number of residue islands). While RFdiffusion performs marginally better on atomic motifs with one residue island, RFdiffusion2 is able to solve complex scaffolding problems with up to seven residue islands. c, In silico success rate across each case for RFdiffusion2 and RFdiffusion. RFdiffusion2 samples at least one scaffold passing the success filters for 41/41 cases while RFdiffusion achieves 16/41, and RFdiffusion2 has a higher in silico success rate than RFdiffusion on all but one case. Difficulty correlates with more residue islands. d, Left, Distribution of TM scores to the closest structure in the PDB for successful in silico designs. Most designs have low closest TM scores between 0.5 and 0.6. Right, A representative superposition of an RFdiffusion2 design (colored) on the closest PDB structure (in gray). e, Comparison of the three approaches for rotamer and index selection for representative AME benchmark cases from each of the residue island categories—3, 4, 5 and 6. One hundred designs were generated for each motif using each approach. Here we display the total in silico success rate over all residue island categories, while in Supplementary Table 1 we show the success rate per category. ‘Inferred’ indicates that RFdiffusion2 generates the rotamer or index during the flow-matching trajectory. ‘Native’ indicates that RFdiffusion2 is given the native rotamer or index at input. ‘Naive’ indicates that RFdiffusion2, as with RFdiffusion, is provided with randomly selected side-chain rotamers and residue indices. We find that ‘naive’ results in the worst overall in silico success rate—as expected, because RFdiffusion fails in most cases. Using native results in more in silico successes, as expected, since the native rotamers and residue spacings are the result of evolutionary optimization. The best performance is achieved by allowing RFdiffusion2 to infer both rotamers and indices, which provides a far larger space to sample over to find optimal solutions than the other two approaches.
We evaluate RFdiffusion and RFdiffusion2 by sampling 100 structures for each benchmark case with the atomic motif as conditioning information. We assign eight sequences to each structure with LigandMPNN32, conditioned on the backbone, catalytic residue side-chain and ligand coordinates. To evaluate whether the sequences fold to the intended structures, we use Chai-1 (ref. 33), an open-source implementation of AlphaFold3 (ref. 34), for structure prediction rather than AlphaFold2 (ref. 35) because we find that Chai-1’s side-chain interaction distances more closely align with the reference distribution of native side chains (Supplementary Fig. 2). We call a design an in silico success if (1) the root mean square deviation of all the heavy atoms in the catalytic residues is <1.5 Å when aligned on the backbone N, Cα, C of those catalytic residues in a Chai-1 prediction for at least one of the LigandMPNN sequences, and (2) the design contains no clashes with the ligand where a clash is defined as two atoms being within 1.5 Å. We release this benchmark that we call atomic motif enzyme (AME) for the scientific community.
To our knowledge, RFdiffusion is the only deep learning method that has been shown to successfully design de novo enzymes13,36. We compare RFdiffusion2 to RFdiffusion by establishing a pipeline that processes each atomic motif into a suitable input for RFdiffusion. The pipeline samples inverse rotamers and residue indices for the atomic motif to transform it into an indexed, backbone motif, and replaces the ligand with an attractive–repulsive potential. RFdiffusion2 generates enzymes conditioned directly on the theozyme without additional processing (Fig. 2a).
We find that RFdiffusion2 finds solutions to all 41 benchmark cases while RFdiffusion finds solutions for only 16/41 cases. In 40/41 cases, we find that RFdiffusion2 substantially outperforms RFdiffusion, setting a new state of the art for theozyme scaffolding (Fig. 3c). We find the difficulty of a benchmark case correlates with the complexity of the motif, which we quantify with the number of ‘residue islands’, the number of contiguous segments of catalytic residues in the original PDB structure. The in silico successes from RFdiffusion2 are quite different from any protein in the training set as measured by FoldSeek37 and template modeling (TM) score38 (Fig. 3d). Although the motif examples in AME come from the PDB, RFdiffusion2 is able to find completely new scaffolds that house these motifs.
We sought to understand the relative contribution of atomic motif and unindexed motif scaffolding to the improved in silico success rates of RFdiffusion2. To resolve the rotamers of the atomic motif, we compare three approaches: naive inverse rotamer sampling as done with RFdiffusion, inferring the rotamer with RFdiffusion2, and the reference case of using the rotamer present in the native structure. For specifying the index of the atomic motif, we compare combinatorial index sampling prior to generative modeling, as done in RFdiffusion, RFdiffusion2 inference of the index, and the reference case of using the index present in the native structure. We evaluated every combination of the settings over four cases in the AME benchmark where each case is randomly chosen from 3, 4, 5 and 6 residue islands. We find the best strategy is to infer both the rotamer and sequence indices—even surpassing using the native rotamer and sequence indices, which would not be available when designing enzymes for novel reactions (Fig. 3e). At four residue islands, we find the naive sampling strategy in RFdiffusion fails to achieve any in silico successes (Supplementary Table 1). We analyzed the diversity of the residues surrounding the motif and found the greatest structural diversity with the unindexed atomic motif strategy, followed by atomic motif and, lastly, backbone motifs with the lowest diversity (Supplementary Fig. 4). Our results show a deep learning approach to resolve the additional degrees of freedom represented by the rotamers and sequence indices is more effective than fixing these to specific values or pre-enumeration.
In vitro experiments
We next experimentally tested whether the model was capable of generating functional enzymes from theozymes. For two reactions, we used theozymes from enzyme crystal structures to decouple the problem of theozyme design from theozyme scaffolding and directly assess the model’s capability on the latter. For another three reactions, we assessed whether it was possible to design a functional enzyme starting from only a desired catalytic mechanism, that is, without a priori knowledge of a functional theozyme geometry. For these cases, we perform optimization with density functional theory (DFT) to find the saddle points of the energy landscape corresponding to transition state geometries of each case. In all five cases, we generate structures with RFdiffusion2 from the input theozyme, fit sequences to those structures, and filter them with structure prediction models to select designs for experimental validation. In all cases, we found functional enzymes when testing less than 96 designed proteins. The specifics of theozyme preparation and experimental characterization for these four reactions are described below.
The aldol reaction forms carbon–carbon bonds between two carbonyl reactants. Enzymatic catalysis of this reaction allows for regiochemical and stereochemical control that would be impossible with nonbiological catalysts. Directed evolution campaigns have demonstrated that a catalytic tetrad composed of a nucleophilic lysine enmeshed in a hydrogen-bonding network with two tyrosines and an asparagine can stabilize the transition states of this reaction39. We constructed a minimal theozyme, comprising the hydrogen bond donors and acceptors of this network and the terminal CE and NZ atoms of lysine required to position the NZ for nucleophilic attack on the reactant, from the crystal structure of one such evolved retroaldolase: RA95.5-8F (PDB 5AN7)39. We generated designs scaffolding this theozyme, filtered them and expressed 96 in an in vitro transcription/translation system. We tested activity of the in vitro transcription/translation-produced proteins using racemic Methodol as a substrate and found four variants that show detectable levels of retroaldolase activity in our semiquantitative assay40. We purified the most active design and found it catalyzed the retroaldolase reaction with kcat/KM of 6.34 ± 0.92 M−1s−1 (Fig. 4a).

Columns from left to right are reaction mechanism hypothesis, a theozyme based on this hypothesis input into RFdiffusion2, a resulting design, the closest protein in the training set (by Foldseek) and experimental activity. In kinetics plots (rightmost column), all points represent mean initial rate values, and error bars represent standard errors over three technical replicates. a, Designed retroaldolase. From left to right, Retroaldolase reaction mechanism uses an activated lysine as a nucleophile; input to RFdiffusion2 (taken from PDB 5AN7); design from RFdiffusion2; superimposition with closest structure in the PDB (6SU3; TM score = 0.47), kinetics assay measuring fluoresence of the product (kcat/KM = 6.34 M−1s−1; literature reports Kuncat = 6.5 × 10−9s−1). Measurement error bars indicate two measurements made on different days. kcat/KM error bars indicate error of fit to Michaelis–Menten equation. b, Designed hydrolase using nucleophilic cysteine. From left to right, Reaction mechanism involving a catalytic triad with a cysteine and an oxyanion hole to stabilize the transition state; inputs to the model derived from PDB 1PPN; design from RFdiffusion2; superimposition with closest structure in the PDB (5K7V; TM score = 0.53); kinetics assay measuring a fluorescence of the product once hydrolyzed (kcat/KM = 250 M−1s−1, Kuncat = 9 × 10−6s−1 shown in Supplementary Fig. 15). c, Designed hydrolase using zinc as a Lewis acid and 4MU-butyrate as a reactant. From left to right, Reaction mechanism involving an activated water molecule as a nucleophile; DFT computed side-chain coordinates that position the zinc and substrate in the optimal geometry for the reaction; design from RFdiffusion2; superimposition with closest structure in the PDB (2ZTK; TM score = 0.54); kinetics assay performed in the same manner as in b (kcat/KM = 77 M−1s−1, Kuncat = 9 × 10−6s−1 shown in Supplementary Fig. 15). d, Designed hydrolase using zinc as a Lewis acid and 4MU-phenylacetate as a reactant. From left to right, Reaction mechanism involving an activated water molecule as a nucleophile; DFT computed side-chain coordinates that position the zinc and substrate in the optimal geometry for the reaction; design from RFdiffusion2; superimposition with closest structure in the PDB (4WIB; TM score = 0.5); kinetics assay performed in the same manner as in b and c (kcat/KM = 16, 000 M−1s−1, Kuncat = 2.1 × 10−5s−1 shown in Supplementary Fig. 15). e, Designed hydrolase using zinc as a Lewis acid and 4MU-phenylacetate as a reactant, and glutatmate as a general base. From left to right, Reaction mechanism involving a glutamate activating a water molecule, which will serve as a nucleophile; DFT computed side-chain coordinates that position the zinc and substrate in the optimal geometry for the reaction; design from RFdiffusion2; superimposition with closest structure in the PDB (2EI4; TM score = 0.60); kinetics assay performed in the same manner as in b and c (kcat/KM = 53, 000 M−1s−1, Kuncat = 2.1 × 10−5s−1 shown in Supplementary Fig. 15).
Esterases catalyzing the cleave of an ester bond by water perform many cellular functions41 and have numerous industrial applications42,43. As a first route to ester cleavage, we chose a cysteine hydrolase theozyme consisting of a Cys-His-Asn catalytic triad in which the cysteine performs nucleophilic attack, with the histidine acting as a general acid/base activated by the asparagine, and a helix-dipole-stabilized oxyanion hole formed by the cysteine backbone nitrogen together with a glutamine that stabilizes the negative charge of the tetrahedral intermediate formed during nucleophilic addition. We take as the minimal catalytic components for this reaction the active functional groups of the cysteine, histidine, asparagine and glutamine, and 3–4 backbone atoms of the residues abutting the cysteine to force the local backbone into a region of Ramachandran space corresponding to the termination of a helix oriented in such a way that its dipole stabilizes the local oxyanion hole. We took the relative positions of these atoms from the crystal structure of a papaya cysteine hydrolase (PDB 1PPN)44, positioned our chosen substrate 4MU-butyrate according to the known optimal geometries for nucleophilic attack by the cysteine, proton donation by the histidine, and hydrogen bond stabilization of the oxyanion45. Among the 48 designs that were screened experimentally, several displayed detectable activity (Supplementary Fig. 12); the best design shown exhibited multiple turnover activity with a kcat/KM of 248 ± 34 M−1s−1 for the acylation step, better than previous results for designed cysteine esterases46 for the same leaving group (Fig. 4b).
Metallohydrolases coordinate metal ions and leverage their Lewis acidity to activate water to form a potent nucleophile capable of hydrolyzing some of the most stable molecules in biological systems. Harnessing this mechanism for arbitrary substrates would enable the design of de novo enzymes capable of hydrolyzing environmental pollutants with long half-lives47,48. We used RFdiffusion2 to design metallohydrolases for two substrates (4MU-butyrate and 4MU-phenylacetate) as described in detail in the accompanying paper49; here we provide a brief summary of the design strategy and experimental results. In contrast to the previous case studies in which the theozyme geometry was extracted from native enzymes, to design the theozyme, we used DFT to find the geometry of the transition state in which the hydroxide ion forms a bond with the carbonyl carbon, simulating the Zn(II) metal, metal-coordinating functional groups (imidazole), a chosen reactant and a hydroxide ion. We obtained 96 designs for each and identified three functional enzymes for the 4MU-butyrate reaction and five for the 4MU-phenylacetate reaction. We found the best enzyme for the 4MU-butyrate reactant had a kcat/KM of 77 ± 10 M−1s−1, higher than previously designed zinc hydrolases50,51. The best enzyme for 4MU-phenylacetate had a kcat/KM of 16,000 ± 2,000 M−1s−1, several orders of magnitude higher than previously designed zinc hydrolases. In a second set of 96 designs tested including a general base to activate the water, we identified 11 functional enzymes, the best of which had a kcat/KM of 53,000 ± 5,000 M−1s−1 (Fig. 4c–e). For more details, see ref. 49.
These experimental results demonstrate that RFDiffusion2 is able to generate functional enzymes when screening less than 96 designs both by scaffolding theozymes from native enzymes and from DFT calculations. The most active design for each reaction is structurally distinct from all structures in the PDB (Fig. 4; Novelty column).
Discussion
RFdiffusion2 outperforms the prior state of the art methods on in silico benchmarks, removes expert intuition necessary with prior backbone motif-scaffolding and scaffold library methods, and can design enzymes with considerable experimentally confirmed catalytic activity. RFdiffusion2 enables direct scaffolding of ideal active sites described at the atom level without prespecifying sequence indices or enumerating side-chain rotamers. We show on our newly curated AME benchmark that RFdiffusion2 substantially improves on RFdiffusion over a range of atom-level active site descriptions. Our design campaigns for retroaldolases, cysteine hydrolases and zinc hydrolases found active and novel enzymes for each reaction. Our in silico success in the AME benchmark suggests RFdiffusion2 should be applicable to designing enzymes across many more reactions at higher success rates than the prior state of the art.
There are several avenues for improvement. Despite RFdiffusion2’s success in obtaining active enzymes across four reactions, the enzymes designed by RFdiffusion2 are not as active as native enzymes. Our theozymes might not be capturing all the necessary interactions for high activity and RFdiffusion2 might be able to sample higher-activity enzymes by expanding our theozyme definition to include more interactions that are necessary for catalysis13. Automating the design of enzymes from theozymes opens up the possibility of large-scale testing of varied theozymes to broaden our understanding of enzymes and validate mechanistic hypotheses of much wider scope than those testable with catalytic residue knockout experiments or directed evolution. Alternative neural network architectures such as Diffusion Transformers52 and modules from AlphaFold3 (ref. 34) for all-atom tasks could improve RFdiffusion2, which uses the neural network architecture of RFAA. The AME benchmark is limited to scaffolding motifs in the PDB derived from annotations of native enzymes from M-CSA. Although beyond the scope of our work, extending AME to measure success on enzymes with multiple transition states could help advance de novo enzyme design. As more successful theozymes from DFT are validated, we expect it will become possible to benchmark non-PDB motifs in the near future. The rapid development of open-source biomolecular interaction structure prediction methods such as Chai-1, Boltz-1 (ref. 53), Protenix54 and ESMFold55 could lead to improved filtering of successful enzyme designs. Finally, we expect that co-designing the protein sequence10 and side chains56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71 outside the active site could lead to more favorable pocket interactions with the substrate and potentially enable sequence-based guidance based on experimental kinetics data.
RFdiffusion2 should be immediately useful to protein designers working on design problems requiring atomic-resolution modeling such as small-molecule binding and enzyme design. We expect that the introduction of RFdiffusion2 and the AME benchmark will open up new research efforts in the machine learning community exploring the design of new modeling approaches for atomic-resolution protein design. To this end, we are making the RFdiffusion2 code freely available to the research community.
Methods
A detailed description of the methods is provided in the Supplementary Information. A brief summary is provided below. Code to run RFdiffusion2 and reproduce the results of the paper are available on https://github.com/RosettaCommons/RFdiffusion2/.
Architecture and training
RFdiffusion2 builds on the representation of previous motif-scaffolding methods9 by adding (1) a new class of conditioning inputs that are represented at the atomic level (as opposed to backbone motifs in previous methods) and (2) a class of motifs for which the residue index is not shown. To handle this, atomic and unindexed motifs are provided as in-context conditioning to the RF All-Atom network architecture18. The network is trained with a flow-matching objective derived in ref. 24. There are several differences to the original RFdiffusion: (1) RFdiffusion2 can handle unindexed and atomic motifs including small molecules, (2) the network is trained from random initialization instead of fine-tuned from structure prediction weights, and (3) the network uses a flow-matching objective instead of a diffusion objective resulting in more stable training. Further details are provided in the Supplementary Methods (section B).
AME benchmark
To our knowledge, a previous benchmark for unindexed atomic motif scaffolding has not been constructed. We developed a benchmark by taking all enzymes in the M-CSA30, cross-correlating them to find structures where all reactants were present using the PARITY database31. After applying final quality filters (described in Supplementary Methods), we found 41 cases that satisfy all the criteria. For each catalytic residue labeled in the M-CSA, we chose a random subgraph of the residue to be the catalytic atoms. It is important to note that scaffolding these atoms will not necessarily generate catalytically proficient enzymes, but these cases were extremely helpful in evaluating the ability of the network to perform unindexed atomic motif scaffolding.
RFdiffusion2 is run with all the default configurations, placing the origin token at the center of mass of the atomic motif. RFdiffusion1 is run as a baseline by randomly sampling indices, using inverse rotamer sampling to sample plausible backbone positions for the atomic motif, and using a substrate repulsive potential to create a pocket for the ligand. For each case, we generated 100 backbones, fit eight sequences to them with LigandMPNN32 and then refolded them with Chai-1 (ref. 33). A successful backbone has at least one sequence where one of the five Chai-1 diffusion samples has all the atomic side chains within 1.5 Å when the backbones of the output generation motif residues are aligned to the predicted backbone coordinates and having no ligand clashes with backbone atoms in the prediction. Further details are provided in the Supplementary Methods (section B).
Experimental characterization of enzymes
The design procedure for all design campaigns was to select an input theozyme, generate backbones with RFdiffusion2, assign multiple sequences using LigandMPNN, refold with a structure prediction network such as AF2 or Chai-1 and apply problem specific filters to select designs to order. Experimental characterization involved obtaining synthetic genes for designs, expressing and purifying the proteins and measuring initial rates at different concentrations to determine Michaelis–Menten kinetics. We describe the specifics of each design campaign in the Supplementary Methods. Details of each design campaign and experimental characterization are described in the Supplementary Methods (section D).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
PDB structures used for training and evaluation were obtained from the RCSB. Enzyme information and metadata are available in the publicly available M-CSA database. Source data are provided with this paper.
Code availability
Code for running RFdiffusion2 is available under the MIT open-source license on GitHub via https://github.com/RosettaCommons/RFdiffusion2/.
References
Lovelock, S. L. et al. The road to fully programmable protein catalysis. Nature 606, 49–58 (2022).
Hossack, E. J., Hardy, F. J. & Green, A. P. Building enzymes through design and evolution. ACS Catalysis 13, 12436–12444 (2023).
Yeh, A. H.-W. et al. De novo design of luciferases using deep learning. Nature 614, 774–780 (2023).
Zanghellini, A. et al. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 15, 2785–2794 (2006).
Song, Y. et al. Score-based generative modeling through stochastic differential equations. In International Conference on Learning Representations (2021).
Ho, J., Jain, A. & Abbeel, P. Denoising diffusion probabilistic models. Adv. Neural Inf. Proc. Syst. 33, 6840–6851 (2020).
Lipman, Y., Chen, R. T., Ben-Hamu, H., Nickel, M. & Le, M. Flow matching for generative modeling. In International Conference on Learning Representations (2023).
Albergo, M. S., Boffi, N. M. & Vanden-Eijnden, E. Stochastic interpolants: a unifying framework for flows and diffusions. In International Conference on Learning Representations (2023).
Watson, J. L. et al. De novo design of protein structure and function with RFdiffusion. Nature 620, 1089–1100 (2023).
Campbell, A., Yim, J., Barzilay, R., Rainforth, T. & Jaakkola, T. Generative flows on discrete state-spaces: enabling multimodal flows with applications to protein co-design. In Proceedings of the 41st International Conference on Machine Learning 5453–5512 (JMLR, 2024).
Lin, Y., Lee, M., Zhang, Z. & AlQuraishi, M. Out of many, one: designing and scaffolding proteins at the scale of the structural universe with Genie 2. Preprint at https://arxiv.org/abs/2405.15489 (2024).
Huguet, G. et al. Sequence-augmented SE (3)-flow matching for conditional protein backbone generation. Adv. Neural Inf. Process. Syst. 38 (2024).
Anna Lauko et al. Computational design of serine hydrolases. Science 388, eadu2454 (2025).
Braun, M. et al. Computational design of highly active de novo enzymes. Preprint at bioRxiv https://doi.org/10.1101/2024.08.02.606416 (2024).
Houk, K. & Liu, F. Holy grails for computational organic chemistry and biochemistry. Acc. Chem. Research 50, 539–543 (2017).
Buller, R. et al. From nature to industry: harnessing enzymes for biocatalysis. Science 382, 8615 (2023).
Reisenbauer, J. C., Sicinski, K. M. & Arnold, F. H. Catalyzing the future: recent advances in chemical synthesis using enzymes. Curr. Opin. Chem. Biol. 83, 102536 (2024).
Krishna, R. et al. Generalized biomolecular modeling and design with RoseTTAFold All-Atom. Science 384, 2528 (2024).
Chen, T., Zhang, R. & Hinton, G. Analog bits: generating discrete data using diffusion models with self-conditioning. In International Conference on Learning Representations (2023).
Esser, P. et al. Scaling rectified flow transformers for high-resolution image synthesis. In Proceedings of the 41st International Conference on Machine Learning 12606–12633 (JMLR, 2024).
Ma, N. et al. SiT: exploring flow and diffusion-based generative models with scalable interpolant transformers. In European Conference on Computer Vision (eds Leonardis, A. et al.) 23-40 (Springer, Cham., 2024).
Liu, X., Gong, C., Liu, Q. Flow straight and fast: learning to generate and transfer data with rectified flow. In International Conference on Learning Representations (2023).
Yim, J. et al. Fast protein backbone generation with SE(3) flow matching. Preprint at https://arxiv.org/abs/2310.05297 (2023).
Yim, J. et al. Improved motif-scaffolding with SE(3) flow matching. Transactions on Machine Learning Research 2712 (2024).
Chen, R. T. & Lipman, Y. Flow matching on general geometries. In International Conference on Learning Representations (2024).
Berman, H. M. et al. The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000).
Tien, M. Z., Meyer, A. G., Sydykova, D. K., Spielman, S. J. & Wilke, C. O. Maximum allowed solvent accessibilites of residues in proteins. PLoS ONE 8, 80635 (2013).
Shrake, A. & Rupley, J. A. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Mol. Biol. 79, 351–371 (1973).
Zheng, Z. et al. MotifBench: a standardized protein design benchmark for motif-scaffolding problems. Preprint at https://arxiv.org/abs/2502.12479 (2025).
Ribeiro, A. J. M. et al. Mechanism and catalytic site atlas (M-CSA): a database of enzyme reaction mechanisms and active sites. Nucleic Acids Res. 46, 618–623 (2018).
Tyzack, J. D., Fernando, L., Ribeiro, A. J., Borkakoti, N. & Thornton, J. M. Ranking enzyme structures in the PDB by bound ligand similarity to biological substrates. Structure 26, 565–571 (2018).
Dauparas, J. et al. Atomic context-conditioned protein sequence design using ligandMPNN. Nat. Methods 22, 717–723 (2025).
Discovery, C. et al. Chai-1: decoding the molecular interactions of life. Preprint at bioRxiv https://doi.org/10.1101/2024.10.10.615955 (2024).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Jiang, L. et al. De novo computational design of retro-aldol enzymes. Science 319, 1387–1391 (2008).
Van Kempen, M. et al. Fast and accurate protein structure search with foldseek. Nat. Biotechnol. 42, 243–246 (2024).
Zhang, Y. & Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 57, 702–710 (2004).
Obexer, R. et al. Emergence of a catalytic tetrad during evolution of a highly active artificial aldolase. Nat. Chem. 9, 50–56 (2017).
Anishchenko, I. et al. Modeling protein-small molecule conformational ensembles with PLACER. Proc. Natl Acad. Sci.USA 122, e2427161122 (2025).
Chapman, H. A., Riese, R. J. & Shi, G.-P. Emerging roles for cysteine proteases in human biology. Annu. Rev. Physiol. 59, 63–88 (1997).
Panke, S., Held, M. & Wubbolts, M. Trends and innovations in industrial biocatalysis for the production of fine chemicals. Curr. Opin. Biotechnol. 15, 272–279 (2004).
Hult, K. & Berglund, P. Engineered enzymes for improved organic synthesis. Curr. Opinion Biotechnol. 14, 395–400 (2003).
Pickersgill, R., Harris, G. & Garman, E. Structure of monoclinic papain at 1.60 Å resolution. Struct. Sci. 48, 59–67 (1992).
Buller, A. R. & Townsend, C. A. Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad. Proc. Natl Acad. Sci. USA 110, 653–661 (2013).
Richter, F. et al. Computational design of catalytic dyads and oxyanion holes for ester hydrolysis. J. Am. Chem. Soc. 134, 16197–16206 (2012).
Ellis, L. D. et al. Chemical and biological catalysis for plastics recycling and upcycling. Nat. Catal. 4, 539–556 (2021).
Chamas, A. et al. Degradation rates of plastics in the environment. ACS Sustain. Chem. Eng. 8, 3494–3511 (2020).
Kim, D. et al. Computational design of metallohydrolases. Nature https://doi.org/10.1038/s41586-025-09746-w (in the press).
Hoffnagle, A. M. & Tezcan, F. A. Atomically accurate design of metalloproteins with predefined coordination geometries. J. Am. Chem. Soc. 145, 14208–14214 (2023).
Der, B. S. et al. Metal-mediated affinity and orientation specificity in a computationally designed protein homodimer. J. Am. Chem. Soc. 134, 375–385 (2012).
Peebles, W. & Xie, S. Scalable diffusion models with transformers. In Proc. IEEE/CVF International Conference on Computer Vision (ICCV) 4172–4182 (2023).
Wohlwend, J. et al. Boltz-1 democratizing biomolecular interaction modeling. Preprint at bioRxiv https://doi.org/10.1101/2024.11.19.624167 (2024).
ByteDance AML AI4Science Team; Chen, X. et al. Protenix-advancing structure prediction through a comprehensive AlphaFold3 reproduction. Preprint at bioRxiv https://doi.org/10.1101/2025.01.08.631967 (2025).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Chu, A. E. et al. An all-atom protein generative model. Proc. Natl Acad. Sci. USA 121, 2311500121 (2024).
McGibbon, R. T. et al. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 109, 1528–1532 (2015).
Lipman, Y. et al. Flow matching guide and code. Preprint at https://arxiv.org/abs/2412.06264 (2024).
Baek, M. et al. Efficient and accurate prediction of protein structure using RoseTTAFold2. Preprint at bioRxiv https://doi.org/10.1101/2023.05.24.542179 (2023).
Kingma, D. P. & Ba, J. Adam: a method for stochastic optimization. In International Conference for Learning Representations (2015).
Kipnis, Y. et al. Design and optimization of enzymatic activity in a de novo β-barrel scaffold. Protein Sci. 31, e4405 (2022).
Althoff, E. A. et al. Robust design and optimization of retroaldol enzymes. Protein Sci. 21, 717–726 (2012).
Tyka, M. D. et al. Alternate states of proteins revealed by detailed energy landscape mapping. J. Mol. Biol. 405, 607–618 (2011).
Wicky, B. et al. Hallucinating symmetric protein assemblies. Science 378, 56–61 (2022).
Studier, F. W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41, 207–234 (2005).
Wang, J. et al. Deep learning methods for designing proteins scaffolding functional sites. Science 377, 387–394 (2022).
Leman, J. K. et al. Macromolecular modeling and design in rosetta: recent methods and frameworks. Nat. Methods 17, 665–680 (2020).
Frisch, M. J. et al. Gaussian 16 Revision C 01. (Gaussian Inc., 2016).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Bannwarth, C. et al. Extended tight-binding quantum chemistry methods. Wiley Interdiscip. Rev. 11, 1493 (2021).
Schmidt, T. G. & Skerra, A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2, 1528–1535 (2007).
Acknowledgements
We thank L. Goldschmidt and K. VanWormer for maintaining the computational and wet lab resources at the Institute for Protein Design, S. Mathis for insightful conversations about conditional generative modeling, J. Watson for valuable discussions on objective stability in generative models, S. Pellock and A. Lauko for their insight on the challenges faced by enzyme designers and J. Butcher for detailed discussion of centering strategies. This work was funded by the National Science Foundation Graduate Research Fellowship Program, grant nos. DGE-2140004 (to S.M.W.) and DGE-2141064 (to J.Y.), The Howard Hughes Medical Institute (to D.B., I.K., Y.K. and B.C.), the Open Philanthropy Project Improving Protein Design Fund (to I.K.), The Bill and Melinda Gates Foundation INV-010680 (to D.K. and W.A.), Gift from Microsoft (GF117374: Microsoft Protein Prediction Research) (to R.K.), the Schmidt Futures Foundation (to S.S.), NSF Expeditions grant (award 1918839: Collaborative Research: Understanding the World Through Code; to J.Y., R.B. and T.S.J.), Machine Learning for Pharmaceutical Discovery and Synthesis (MLPDS) consortium (to J.Y., R.B. and T.S.J.), the Abdul Latif Jameel Clinic for Machine Learning in Health (to J.Y., R.B. and T.S.J.), the DTRA Discovery of Medical Countermeasures Against New and Emerging (DOMANE) threats program (to J.Y., R.B. and T.S.J.), the DARPA Accelerated Molecular Discovery program (to J.Y., R.B. and T.S.J.) and the Sanofi Computational Antibody Design grant (to J.Y., R.B. and T.S.J.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
W.A., D.T. and D.B. conceived the study. W.A., J.Y., D.T., S.S. and H.R.A.-T. contributed to development of RFdiffusion2. W.A. trained all versions of RFdiffusion2 described in the study. W.A., S.S. and R.K. performed in silico benchmarking. W.A., S.S., J.Y., and R.K. analyzed RFdiffusion2 results. S.S., S.M.W., D.K., I.K., Y.K. and B.C. experimentally characterized designs. W.A., J.Y., R.K. and D.B. were responsible for the manuscript writing. S.S., S.M.W., D.K. and I.K. were responsible for writing in vitro details. D.B., R.K., T.S.J. and R.B. offered supervision throughout the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks Anastassis Perrakis, Gunnar Schroeder and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Arunima Singh, in collaboration with the Nature Methods team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Additional information to replicate and understand the RFdiffusion2 system and its experiments described in the main text.; Supplementary Figs. 1–17; description of RFdiffusion2 method; description of experiments; and Supplementary Tables 1 and 6.
Source data
Source Data Fig. 3 (download ZIP )
Csv and txt files to plot all graphs, organized by figure panel (PDB files from AME benchmark and ablation study are available on Zenodo).
Source Data Fig. 4 (download ZIP )
xlsx files for kinetic measurements organized by figure panel (PDB files for active designs are available on Zenodo).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ahern, W., Yim, J., Tischer, D. et al. Atom-level enzyme active site scaffolding using RFdiffusion2. Nat Methods 23, 96–105 (2026). https://doi.org/10.1038/s41592-025-02975-x
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41592-025-02975-x
This article is cited by
-
Computational design of metallohydrolases
Nature (2026)
-
Flow matching meets biology and life science: a survey
npj Artificial Intelligence (2026)
