
Abstract
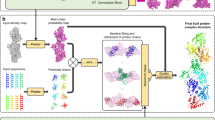
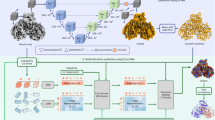
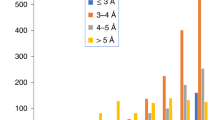
Cryo-electron microscopy (cryo-EM) has become the mainstream technique for macromolecular structure determination. However, because of intrinsic resolution heterogeneity, accurate modeling of all-atom structure from cryo-EM maps remains challenging even for maps at near-atomic resolution. Addressing the challenge, we present EMProt, a fully automated method for accurate protein structure determination from cryo-EM maps by efficiently integrating map information and structure prediction with a three-track attention network. EMProt is extensively evaluated on a diverse test set of 177 experimental cryo-EM maps with up to 54 chains in a case at <4-Å resolution, and compared to state-of-the-art methods including DeepMainmast, ModelAngelo, phenix.dock_and_rebuild and AlphaFold3. It is shown that EMProt greatly outperforms the existing methods in recovering the protein structure and building the complete structure. In addition, the built models by EMrot exhibit a high accuracy in model-to-map fit and structure validations.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others
Data availability
All published datasets used in this paper were taken from the EMDB and PDB (accession codes specified in the figure captions and Supplementary Tables). All raw data of the evaluation results are provided in the article and Supplementary Information. The list of items in the test set is available in Supplementary Table 1. The list of training items is available in Supplementary Table 6. Source data are provided with this paper.
Code availability
The EMProt package is freely available online for academic or noncommercial users (http://huanglab.phys.hust.edu.cn/EMProt or https://github.com/huang-laboratory/EMProt). A link to a demo that runs on Google Colab and requires no installation on the local device is available on the GitHub repository.
References
Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nat. Methods. 13, 24–27 (2016).
Frank, J. Advances in the field of single-particle cryo-electron microscopy over the last decade. Nat. Protoc. 12, 209–212 (2017).
Cheng, Y. Single-particle cryo-EM—how did it get here and where will it go. Science 361, 876–880 (2018).
Lawson, C. L. et al. EMDataBank unified data resource for 3DEM. Nucleic Acids Res. 44, D396–D403 (2016).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in PHENIX. Acta Crystallogr. D Struct. Biol. 75, 861–877 (2019).
De la Rosa-Trevín, J. M. et al. Scipion: a software framework toward integration, reproducibility and validation in 3D electron microscopy. J. Struct. Biol. 195, 93–99 (2016).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods. 14, 290–296 (2017).
Vilas, J. L., Carazo, J. M. & Sorzano, C. O. S. Emerging themes in cryoEM—single particle analysis image processing. Chem. Rev. 122, 13915–13951 (2022).
Berman, H. M. et al. The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000).
Terwilliger, T. C., Adams, P. D., Afonine, P. V. & Sobolev, O. V. A fully automatic method yielding initial models from high-resolution cryo-electron microscopy maps. Nat. Methods. 15, 905–908 (2018).
Terashi, G. & Kihara, D. De novo main-chain modeling for EM maps using MAINMAST. Nat. Commun. 9, 1618 (2018).
He, J. & Huang, S.-Y. Full-length de novo protein structure determination from cryo-EM maps using deep learning. Bioinformatics 37, 3480–3490 (2021).
Si, D. et al. Deep learning to predict protein backbone structure from high-resolution cryo-em density maps. Sci. Rep. 10, 4282 (2020).
Xu, K., Wang, Z., Shi, J., Li, H. & Zhang, Q. C. A2-net: molecular structure estimation from cryo-EM density volumes. In Proc. 33rd AAAI Conference on Artificial Intelligence (eds van Hentenryck, P. & Zhou, Z.-H.) (AAAI, 2019).
Pfab, J., Phan, N. M. & Si, D. DeepTracer for fast de novo cryo-EM protein structure modeling and special studies on CoV-related complexes. Proc. Natl Acad. Sci. USA 118, e2017525118 (2021).
Giri, N. & Cheng, J. De novo atomic protein structure modeling for cryoEM density maps using 3D transformer and HMM. Nat. Commun. 15, 5511 (2024).
Jamali, K. et al. Automated model building and protein identification in cryo-EM maps. Nature 628, 450–457 (2024).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Kawabata, T. Multiple subunit fitting into a low-resolution density map of a macromolecular complex using a Gaussian mixture model. Biophys. J. 95, 4643–4658 (2008).
Wriggers, W. Conventions and workflows for using Situs. Acta Crystallogr. D Biol. Crystallogr. 68, 344–351 (2012).
Kovacs, J. A., Galkin, V. E. & Wriggers, W. Accurate flexible refinement of atomic models against medium-resolution cryo-EM maps using damped dynamics. BMC Struct. Biol. 18, 12 (2018).
He, J., Lin, P., Chen, J., Cao, H. & Huang, S.-Y. Model building of protein complexes from intermediate-resolution cryo-EM maps with deep learning-guided automatic assembly. Nat. Commun. 13, 4066 (2022).
Zhou, X. et al. Progressive assembly of multi-domain protein structures from cryo-EM density maps. Nat. Comput. Sci. 2, 265–275 (2022).
Zhang, Z. et al. DEMO-EM2: assembling protein complex structures from cryo-EM maps through intertwined chain and domain fitting. Brief. Bioinform. 25, bbae113 (2024).
Terwilliger, T. C. et al. Improved AlphaFold modeling with implicit experimental information. Nat. Methods. 19, 1376–1382 (2022).
Zhang, X., Zhang, B., Freddolino, P. L. & Zhang, Y. CR-I-TASSER: assemble protein structures from cryo-EM density maps using deep convolutional neural networks. Nat. Methods. 19, 195–204 (2022).
Terashi, G., Wang, X., Prasad, D., Nakamura, T. & Kihara, D. DeepMainmast: integrated protocol of protein structure modeling for cryo-EM with deep learning and structure prediction. Nat. Methods. 21, 122–131 (2024).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Krogh, A., Brown, M., Mian, I. S., Sjölander, K. & Haussler, D. Hidden Markov models in computational biology. Applications to protein modeling. J. Mol. Biol. 235, 1501–1531 (1994).
Larralde, M. & Zeller, G. PyHMMER: a Python library binding to HMMER for efficient sequence analysis. Bioinformatics 39, btad214 (2023).
Zhang, Y. & Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 (2005).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 74, 531–544 (2018).
Zhang, C., Shine, M., Pyle, A. M. & Zhang, Y. US-align: universal structure alignments of proteins, nucleic acids, and macromolecular complexes. Nat. Methods 19, 1109–1115 (2022).
Afonine, P. V. et al. New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr. D Struct. Biol. 74, 814–840 (2018).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 6, 12–21 (2010).
Kabsch, W. & Sander, C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Zhang, K. et al. Practical blind image denoising via Swin-Conv-UNet and data synthesis. Mach. Intell. Res. 20, 822–836 (2023).
Liu, Z. et al. Swin transformer: hierarchical vision transformer using shifted windows. In Proc. of the IEEE/CVF International Conference on Computer Vision (ed. O’Conner, L.) (IEEE, 2021).
Li, T. et al. All-atom RNA structure determination from cryo-EM maps. Nat. Biotechnol. 43, 97–105 (2025).
Li, T., Cao, H., He, J. & Huang, S.-Y. Automated detection and de novo structure modeling of nucleic acids from cryo-EM maps. Nat. Commun. 15, 9367 (2024).
DiMaio, F., Tyka, M. D., Baker, M. L., Chiu, W. & Baker, D. Refinement of protein structures into low-resolution density maps using Rosetta. J. Mol. Biol. 392, 181–190 (2009).
He, J., Li, T. & Huang, S.-Y. Improvement of cryo-EM maps by simultaneous local and non-local deep learning. Nat. Commun. 14, 3217 (2023).
Wang, Z., Bovik, A. C., Sheikh, H. R. & Simoncelli, E. P. Image quality assessment: from error visibility to structural similarity. IEEE Trans. Image Process. 13, 600–612 (2004).
Zhu, K., Su, H., Peng, Z. & Yang, J. A unified approach to protein domain parsing with inter-residue distance matrix. Bioinformatics 39, btad070 (2023).
Terwilliger, T. C. et al. AlphaFold predictions are valuable hypotheses and accelerate but do not replace experimental structure determination. Nat. Methods. 21, 110–116 (2024).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (32161133002, 32430020 and 62072199 to S.-Y.H.), the startup grant of Huazhong University of Science and Technology (to S.-Y.H.) and the Postdoctoral Fellowship Program of the China Postdoctoral Science Fund (GZB20250617 to T.L.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
S.-Y.H. conceptualized and supervised the project. T.L., J.C., H.L. and H.C. designed and performed the experiments. S.-Y.H. and T.L analyzed the data. T.L. and S.-Y.H. wrote the paper. All authors reviewed and approved the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Structural & Molecular Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Sara Osman, in collaboration with the Nature Structural & Molecular Biology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–4.
Supplementary Tables
Supplementary Tables 1–6.
Source data
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, T., Chen, J., Li, H. et al. EMProt improves structure determination from cryo-EM maps. Nat Struct Mol Biol (2025). https://doi.org/10.1038/s41594-025-01723-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41594-025-01723-1
This article is cited by
-
When cryo-EM modeling meets structure prediction
Nature Structural & Molecular Biology (2026)
