
Abstract
Bitter taste functions as a means of both protection against potentially toxic compounds and savoring bitter tasting foods and beverages. Among the 26 bitter taste receptors, taste receptor type 2 member 43 (TAS2R43) has been identified as key for recognizing the bitter taste of coffee. TAS2R43 has also been implicated in many other physiological processes, including the regulation of glucagon-like peptide 1 release from the intestine, bronchodilation, innate immunity and metabolism. Here we report cryo-electron microscopy structures of human TAS2R43 coupled with inhibitory G protein or gustducin (Ggust) stabilized by the potent nephrotoxin and carcinogen aristolochic acid I. Both structures revealed that aristolochic acid I binds in a presumed orthosteric pocket shared with other bitter taste receptor. Further structural, functional and computational studies revealed potential modes for coffee’s constituents including caffeine and cafestol, which are bitter tastants from coffee. Lastly, long-timescale molecular dynamics simulations identified potential cryptic allosteric pockets in TAS2R43. These structures could accelerate the search for specific bitter taste ligands that ultimately may be therapeutically useful.
Similar content being viewed by others

Main
Humans sense five basic tastes: sweet, umami, bitter, sour and salty1,2,3,4. Sweet and umami tastes facilitate the preference for consuming carbohydrates and proteins, respectively, while salty taste contributes to maintaining sodium balance in the body1. By contrast, bitter and sour tastes help humans avoid potentially harmful chemicals and spoiled or fermented foods1,3,4 and to identify foods with potentially flavorful tastes.
Unlike sour and salty taste, which are mediated by ion channels, umami, sweet and bitter taste sensations are mediated by the family T (taste) G-protein-coupled receptors (GPCRs)1,5. Sweet and umami receptors (taste receptor type 1; TAS1R or T1R) are heterodimeric class C GPCRs, composed of TAS1R2 + TAS1R3 and TAS1R1 + TAS1R3 dimers, respectively6. By contrast, 26 family T bitter taste GPCRs (also known as taste receptor type 2; TAS2R or T2R) recognize bitterness and, although structurally similar to class A GPCRs, lack certain conserved structural motifs7. TAS2Rs are expressed in taste receptor cells in the tongue, where they couple to gustducin (Ggust), thereby mediating a complex signaling pathway that ultimately communicates the perception of bitterness8. Additionally, many studies have demonstrated TAS2R expression in extraoral tissues, where they are involved in many physiological functions beyond taste perception, presumably mediated by inhibitory G protein (Gi)9 coupling.
TAS2Rs recognize various chemically diverse compounds, exhibiting potencies with half-maximal response (EC50) values ranging from micromolar to millimolar in cell-based functional assays10. Interestingly, previous studies suggest that several TAS2Rs (TAS2R14, TAS2R31, TAS2R40, TAS2R41 and TAS2R43) are potently activated by (EC50 ranging from nanomolar to micromolar range) aristolochic acid I (AAI), a highly toxic and bitter alkaloid identified as a nephrotoxin and carcinogen extracted from members of the Aristolochiaceae family10. Among these, TAS2R43, which is responsible for sensing the bitter taste of coffee ingredients including caffeine11,12, exhibited the highest affinity for AAI, a heterocyclic aromatic compound structurally similar to purines10. Additionally, previous studies have suggested physiological roles for extraoral TAS2R signaling including TAS2R43 in intestinal glucagon-like peptide 1 release, bronchodilation, innate immunity and metabolism13,14,15,16,17.
Interestingly, a previous study revealed that caffeine activates several bitter taste receptors, including TAS2R7, TAS2R10, TAS2R14, TAS2R43 and TAS2R46 (ref. 18). Beyond caffeine, coffee’s bitterness is proposed to be perceived through TAS2R43 and TAS2R46, which are more selectively activated by mozambioside, cafestol, kahweol and bengalensol, as recently reported19,20. Mozambioside is a precursor of cafestol, kahweol and bengalensol, sharing an ent-kaurane diterpenoid structure with those three compounds, and was predicted as achieved binding at the orthosteric pocket of TAS2R43 using a homology-model-based docking19. Additionally, several other compounds including trigonelline, chlorogenic acids, chlorogenic acid lactones, catechol and proline-based diketopiperazines have been reported as coffee constituents21,22,23,24,25,26, although whether these compounds act as agonists at TAS2R43 or TAS2R46 remains unknown.
Recently, miniGs/gust-coupled TAS2R46 structures bound to strychnine were reported27. These structures revealed that strychnine binds to a presumably orthosteric pocket located in a similar position as that seen in many class A GPCRs. This study also revealed key mechanisms for ligand recognition and for specifying the constitutive activity of TAS2R46 (ref. 27). By contrast, we recently reported the structures of TAS2R14 coupled to Gi and Ggust and the bitter tastant cmpd28.1. Surprisingly, we found an endogenous cholesterol molecule bound to the orthosteric pocket, while cmpd28.1 bound to a previously uncharacterized intracellular site, mediating interactions between the receptor and the helix α5 of the Gα subunit28. This intracellular site for bitter tastants was shortly thereafter also reported by three other independent groups29,30,31. Collectively, these studies revealed how the bitter taste receptor TAS2R14 recognizes chemically diverse ligands, including sterols and anthranilic acid derivatives, and suggested that this intracellular site facilitates direct binding of intracellular tastants with both TAS2R14 and G proteins28,29,30,31. The intracellular site was also engaged by AAI in a similar binding mode to cmpd28.1 in one of these studies29. Previous studies revealed that AAI selectively activates TAS2R31 and TAS2R43 but not TAS2R46 (ref. 32), even though these receptors share common agonists such as lactucin and its metabolites33, as well as coffee’s ingredients19,20 and antagonists such as citronellal11.
However, currently, we lack structural information clarifying how other bitter taste receptors recognize toxic compounds such as AAI, which is recognized by many TAS2Rs10, as well as how coffee’s bitterness is mediated by bitter taste receptors at the molecular level. Here, we present two cryo-electron microscopy (cryo-EM) structures of AAI-bound TAS2R43 in complex with either Gαi1 or gustducin. AAI was observed in a canonical orthosteric pocket with a similar location to the cholesterol-binding site of TAS2R14. Our structures reveal how the bitter taste receptor TAS2R43, which responds to coffee’s ingredients including caffeine, mozambioside and other bitter substituents20, recognizes AAI distinctly from other reported TAS2Rs. Lastly, our molecular dynamics (MD) simulation revealed potential cryptic pockets and computational molecular docking suggested binding modes against diverse compounds.
Results
Structures of TAS2R43–Gi1 and TAS2R43–Ggust complexes
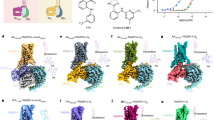
For structure determination, TAS2R43 and either Gαi1β1γ2 or Gαgustβ1γ2 were coexpressed in Spodoptera frugiperda 9 (Sf9) cells as previously described28. To enhance the expression of TAS2R43, we fused hemagglutinin (HA) and b562RIL (BRIL) to the N terminus of TAS2R43. To obtain a stable complex, dominant negative Gα subunits were used together with the NanoBiT tethering strategy34, which was introduced to TAS2R43 along with Gαi1β1γ2 and Gαgustβ1γ2 heterotrimers as reported previously28. The potent agonist AAI was added during protein purification and a homogeneous and stable complex was successfully obtained. Eventually, we determined the AAI-bound complex structures of TAS2R43–Gi1–scFv16 and TAS2R43–Ggust–scFv16 at nominal resolutions of 2.9 Å and 3.0 Å for the global maps, 3.1 Å and 3.2 Å for the locally refined maps of TAS2R43 and 2.8 Å and 2.9 Å for the locally refined map of the G proteins with scFv16, respectively, using cryo-EM (Fig. 1a,b, Extended Data Figs. 1 and 2 and Extended Data Table 1). On the basis of these well-resolved maps, we confidently built models of AAI and TAS2R43, except for disordered regions, including the N-terminal region (1–4), extracellular loop 2 (ECL2; 156–165) and C-terminal region (299–309) (Extended Data Fig. 3). In addition, we performed computational analysis using GemSpot software35 to validate the ligand placement in both structures. The computational analysis revealed that the docked AAI models were accurately built and fit into the cryo-EM densities (Extended Data Fig. 4a,b).

a,b, Representative 3D reconstruction maps and models for TAS2R43–Gi1–scFv16 (a) and TAS2R43–Ggust–scFv16 (b) (TAS2R43, orange; Gαi1, purple; Gαgust, pink; Gβ1, sky blue; Gγ2, yellow; scFv16, gray). Modeled AAs observed in the orthosteric binding pockets are highlighted as magenta sticks with the cryo-EM maps in the black boxes.
Our TAS2R43 structures exhibited similar overall topology to previously reported structures of TAS2R14 and TAS2R46 (refs. 27,28,29,36), including the membrane-parallel intracellular loop 2 (ICL2) forming a short helix (Extended Data Fig. 4c). However, distinct differences were observed in the short helix in the ICL2 and transmembrane domain 4 (TM4), exhibiting distances of 8.8 Å (measured between T111 of TAS2R16 and N115 of TAS2R43) and 5.4 Å (measured between P127 of TAS2R16 and L130 of TAS2R43) from those of TAS2R43, respectively. Additionally, an extended TM5 of TAS2R43 reaching toward the extracellular region was revealed, which is distinct from previously reported structures of TAS2R14, TAS2R16 and TAS2R46 (refs. 27,28,29,36) (Extended Data Fig. 4d). Compared to the recent TAS2R14 structure, in which AAI binds to an intracellular site29, AAI was clearly observed in the orthosteric binding pocket in TAS2R43 (Extended Data Fig. 4e).
AAI binds to the orthosteric binding pocket
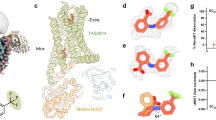
Overall, the two structures of TAS2R43 in complex with either Gi or Ggust are nearly identical, with a root-mean-square deviation (r.m.s.d.) value of 0.5 Å for 245 Cα atoms of TAS2R43 (Extended Data Fig. 5a). The AAI density was observed in the orthosteric binding pocket of both TAS2R43 structures with Gi and Ggust, exhibiting almost identical AAI binding and orientations of the side chains involving AAI binding. Specifically, our structures exhibited that AAI interacts with residues on TM2, TM3, TM6 and TM7 (Fig. 2a,b). W883.32 forms a π–π interaction with the aromatic rings of AAI (Fig. 2c and Extended Data Fig. 5a). A843.28, Y853.29 and F2526.59 form additional hydrophobic interactions with AAI. In particular, F2526.59 in the ECL3 is positioned directly on the extracellular side of AAI, suggesting a potential role as a lid for ligand binding (Fig. 2b,c and Extended Data Fig. 4e). In contrast, S2486.55 forms a hydrogen bond with a nitro group of AAI and R2687.42 forms hydrogen bonds with the nitro group and a carboxyl group. Additionally, R2687.42 forms a specific salt bridge with the carboxyl group with a distance of 2.9 Å.

a, A representative 3D reconstruction model for the Gi-coupled TAS2R43. TAS2R43 and Gαi are colored orange and purple, respectively. A model-fitted map of AAI in the orthosteric pocket is colored magenta. b, Extracellular view of AAI-bound orthosteric binding pocket. F2526.59, a potential lid for ligand sensing, is indicated by a green stick. c, An enlarged view of the AAI-interacting residues indicated by sticks. Hydrogen bonds and ionic interactions are shown as black dashed lines. Models for water molecules are shown as red spheres, with their cryo-EM densities indicated by mesh. d, A dose–response cAMP GloSensor assay stimulated by AAI in HEK293T cells transfected by TAS2R43 of WT and mutant AAI-interacting residues. Data represent the mean ± s.e.m. of n = 3 biological replicates. e,f, AAI-stimulated dose–response BRET2 assays for Gi1 (e) and Ggust (f) using TAS2R43 mutants at the orthosteric pocket. g, Orthogonal views of TAS2R43 representing electrostatic potential surface calculation. Left, same view from a. Red, negative (−5 kT/e); blue, positive (+5 kT/e). AAI is indicated by a magenta stick. h,i, TAS2R43 was activated by coffee compounds in dose–response BRET assays for Gi1 (h) and Ggust (i) using HEK293T cells. The values were normalized to AAI. Data represent the mean ± s.e.m. of n = 4 biological replicates. All of the pEC50 and Emax values for dose–response functional assays are provided in Supplementary Tables 2–4 (d–f,h,i).
Intriguingly, the densities of presumed water molecules were observed inside both TAS2R43 structures in complexes with either Gi or Ggust (Extended Data Fig. 5b,c). In the TAS2R43–Gi complex, the water molecules were observed near N652.60, N76ECL1, W973.41, N1504.63, N1765.40 and A1835.46 and formed hydrogen bonds with those residues (Extended Data Fig. 5b). Specifically, N652.60 formed hydrogen bonds with one of the water molecules (W2) that formed hydrogen bonds with the carboxyl and nitro groups on the extracellular side of AAI (Fig. 2c and Extended Data Fig. 5b). W883.32 and Y2416.48 formed hydrogen bonds with the other molecule (W4), forming hydrogen bonds with the carboxyl group of AAI on the opposite side. By contrast, a distinct distribution of water molecules was observed in the TAS2R43–Ggust complex compared to the TAS2R43–Gi structure. Overall, the water molecules were observed near N652.60, T692.64, R813.26, H1434.56, E1504.63, E1514.64 and S2486.55 and formed hydrogen bonds with those residues in the TAS2R43–Ggust structure (Extended Data Fig. 5c). In detail, at the orthosteric binding pocket, one water molecule (W2) forming a hydrogen bond with the nitro group of AAI was observed on its extracellular side. Additionally, N652.60 formed a hydrogen bond with another water molecule (W3), without directly interacting with AAI. By contrast, no water molecule was observed on the side opposite AAI.
Although we attempted to purify the apo form of TAS2R43 to validate the water networks in the orthosteric pocket, we were unable to obtain a stable complex during purification. Instead, to validate the water densities, we used both phenix.douse and the recently released AI-based prediction tool Metal Ion Classification (MIC)37. The water molecules built by phenix.douse were consistently validated by MIC, supporting their assignments (Supplementary Table 1).
To examine the water molecules in our structures, we then conducted MD simulations using the AAI-bound TAS2R43–Gi structure with the built waters. Overall, the simulations exhibited water densities near all of the water-binding sites in our cryo-EM maps (Extended Data Fig. 5g and Supplementary Fig. 1a). For waters near AAI, the buried W4 site was occupied by one or two water molecules ~50% of the time. In apo simulations without AAI, a single water occupied W4 ~65% of the time, showing a slight shift because of a conformational change of W88 (Extended Data Fig. 5h,i). Given that our TAS2R43–Ggust structure did not show strong water density at this site, the ~50% occupancy at W4 is consistent with placing a water at the corresponding cryo-EM density. By contrast, the upper site (W2), located at the ligand–solvent interface, remained consistently hydrated as expected.
Additionally, to evaluate the role of water in stabilizing AAI binding in the TAS2R43–Gi complex, we conducted two sets of simulations: one retaining the resolved waters and one without them. With the W4 water present, AAI remained stably bound, as W4 persistently bridged between AAI’s carboxylate and the side chains of W88 and Y241 (Extended Data Fig. 5j,k). In contrast, when no water was placed at W4 and this buried pocket did not hydrate within the equilibration timescale, the ligand shifted to an alternative binding mode.
Next, to verify interactions between AAI and the residues in the binding pocket of TAS2R43, we performed bioluminescence resonance energy transfer (BRET2) assays, using our TRUPATH resources38, for Gi1 and Ggust using HEK293T cells transiently transfected with TAS2R43, including the wild type (WT) and N65A, W88A, S248A, F252A and R268A mutants (Fig. 2d,e and Supplementary Table 2). Our assays revealed that the N65A, W88A and R268A substitutions severely impaired TAS2R43 activation for both of Gi1 and Ggust signaling, with a slightly more pronounced impairment in Ggust signaling. Interestingly, the F252A substitution did not affect efficacy and potency of AAI for TAS2R43 activation. Next, we conducted cyclic adenosine monophosphate (cAMP) inhibition assays using the mutant TAS2R43 to evaluate cAMP inhibition levels by the mutants (Fig. 2f and Supplementary Table 3). N65A, W88A and R268A substitutions severely impaired cAMP inhibition levels induced by TAS2R43 activation, consistent with our BRET2 assays, while the S248A substitution reduced activation to half of the maximum activation compared to WT. Interestingly, the F252A substitution slightly enhanced the efficacy and potency of AAI, suggesting a gatekeeping role as a lid against ligand entry, consistent with our structural exhibition (Fig. 2b and Extended Data Fig. 4e).
Our sequence alignment revealed that the AAI-interacting residues are diverse across all TAS2Rs except for W883.32, which is relatively conserved (Extended Data Fig. 6a). Notably, R2687.42 is highly variable, being conserved only between TAS2R43 and TAS2R31, suggesting that it may serve as a key residue sensing AAI, consistent with our functional assays (Fig. 2d–f and Extended Data Fig. 6a). This observation is consistent with a previous mutagenesis study of R268A in TAS2R31 and A268R in TAS2R46, suggesting that R268 is required for AAI sensing32. Our measurement of surface expression levels revealed that a ~45% lower expression of the S248A mutant compared to WT, which may affect AAI-stimulated receptor activation (Extended Data Fig. 8d). By contrast, the R268A substitution severely affected activation despite a ~2.3-fold higher expression compared to WT.
Next, we calculated the electrostatic potential surface of TAS2R43 coupled to Gi to further investigate the orthosteric binding pocket (Fig. 2e). Our studies revealed that the orthosteric binding pocket is highly positively charged, with potential contributions by basic residues K2657.39 and R2687.42 (Fig. 2g). This electrostatic feature may facilitate the recognition of acidic chemicals or negatively charged ligands that are enriched in carboxyl groups like AAI (Fig. 1 and Extended Data Fig. 9). This potential role of the identified residues in sensing ligands, including that of AAI, is consistent with previous mutagenesis studies7,32,39.
Coffee compounds activate TAS2R43
TAS2R43 responds to coffee’s constituents including mozambioside and its metabolites, in addition to caffeine, as previously reported19,20. To examine TAS2R43’s role in coffee bitterness, we conducted BRET2 assays with Gi1 and Ggust using reported coffee compounds, including caffeine, cafestol, catechol, kahweol and trigonelline (Fig. 2h,i and Supplementary Table 4). In the assays, all compounds activated TAS2R43 for Gi1 and, overall, showed lower potencies and efficacies than AAI except for catechol and chlorogenic acid. Compared to AAI, cafestol and kahweol had maximum response (Emax) values greater than 50%, albeit with low EC50 values, whereas catechol activated TAS2R43 for Gi1 with similar EC50 and low Emax values. In contrast, all of the compounds also activated TAS2R43 for Ggust but showed lower potency than for Gi1 activation. Interestingly, our assays revealed that cafestol and kahweol, which are metabolites of mozambioside sharing an ent-kaurane diterpenoid scaffold, activated TAS2R43 for Ggust with efficacies of ~250% and ~150% of AAI, respectively. Collectively, our assay revealed that TAS2R43 detects multiple coffee-derived compounds, including beyond caffeine, cafestol and kahweol, which were previously reported19.
Interactions of TAS2R43 with G proteins
Our structural alignment between the complexes of TAS2R43–Gi and TAS2R43–Ggust revealed that the overall structures are nearly identical, while the G-protein regions exhibited a difference of 3.3 Å between the two αN helices of the Gα subunits (measured at A7) (Fig. 3a). No distinct interaction between the Gα αN helices and TAS2R43 was observed in either complex, despite the 3.3-Å shift. By contrast, a potential hydrogen bond was observed between R38 of the ICL1 of TAS2R43 and D312 of the Gβ subunit in the TAS2R43–Gi structure but not in the TAS2R43–Ggust structure, potentially because of the 1.3-Å shift of Gβ (measured at D312 of Gβ) (Extended Data Fig. 4f–h). Apart from this, these structures revealed similar interactions between TAS2R43 and either Gi or Ggust. The helices α5 of Gα subunits are deeply engaged by the intracellular regions formed by TM2, TM3 and TM5–TM7, as well as ICL2 and ICL3, of TAS2R43. In particular, the tip regions of helices α5 of the Gα subunits (K345 to C terminus) are recognized by TM3 and TM6 (Fig. 3b,d). I1103.54 of TAS2R43 extensively forms hydrophobic interactions with L348, C351 and L353 of the helices α5 of the Gαi and Gαgust. A2276.34 of TAS2R43 formed an additional hydrophobic interaction with L348 of the Gα subunits. Intriguingly, each water molecule density was observed in the interface between the TAS2R43 and the helices α5 of the Gαi and Gαgust, although slightly weaker in the Ggust-coupled TAS2R43 structure, presumably because of its lower resolution. The water molecule mediated hydrogen bonds that formed between Y1063.50 of TAS2R43 and the main chain of C351 of the Gα subunits. Additionally, Y1063.50 formed a hydrophobic interaction with a side chain of the C351. A main chain of K1093.53 of TAS2R43 formed a hydrogen bond with N347 of the helices α5 of the Gαi and Gαgust. Furthermore, K1093.53 formed an ionic interaction with D350 of the helices α5 of the Gαi and Gαgust. To examine the water molecule at the interface between TAS2Rs and the Gα proteins, we compared reported cryo-EM maps of TAS2R14, TAS2R16 and TAS2R46 (refs. 27,28,29,30,31,36). Among these structures, some TAS2R14 maps refined without the deepEMhancer sharpening tool showed a water molecule at the same position between Y1063.50 of TAS2R14 and C351 of the Gα, suggesting that the visibility may depend on cryo-EM data processing and map quality (Extended Data Fig. 5d–f).

a, Orthogonal views for structural superposition of the structures of TAS2R43–Gi–scFv16 (TAS2R43, orange; Gαi, purple; Gβ, sky blue; Gγ, yellow; scFv16, gray) and TAS2R43–Ggust–scFv16 (TAS2R43, brown; Gαgust, pink; Gβ, blue; Gγ, pale red; scFv16, white). A modeled AAI ligands are shown as magenta spheres in the orthosteric pockets. Bottom, the conformational difference in the αN helices is 3.3 Å shown in extracellular view. b, Enlarged view of the boxed region labeled in a, showing the TAS2R43–Gi–scFv16 interface. TAS2R43 residues and Gαi helix α5 residues involved in the interaction are shown as sticks. Hydrogen bonds are indicated by black dashed lines. A water molecule is shown as a red sphere with cryo-EM density (σ = 0.5). c, Orthogonal view of b, highlighting ionic interactions indicated by black dashed lines. d, Identical view to b for the TAS2R43–Ggust–scFv16 complex. A water molecule is shown as a red sphere with cryo-EM density (σ = 0.5). e, Orthogonal view of d. f–i, AAI-stimulated dose–response BRET2 assays for Gi1 (f,g) and Ggust (h,i) using TAS2R43 mutants at the interfaces between TAS2R43 and the Gαi1 and Gαgust. All of the pEC50 and Emax values for dose–response functional assays are provided in Supplementary Tables 5 and 6 (f–i). The values were normalized to WT. Data represent the mean ± s.e.m. of n = 3 biological replicates.
In contrast, the regions of α5 helices of Gαi and Gαgust, closely located to the core of the Gα (T329 to I344), were recognized by the TM5, TM6 and ICL3 of TAS2R43 (Fig. 3c,e). On the interacting regions, the enriched basic residues of TAS2R43 were involved in ionic interactions with the acidic residues of Gαi and Gαgust. Specifically, K2085.71 and H2125.75 of TAS2R43 formed ionic interactions with D337 of the helices α5 of the Gα subunits. H2055.68 and H2246.31 of TAS2R43 form ionic interactions with D341H5.13 of the helices α5 of the Gα subunits. Interestingly, K214 in the ICL3 of TAS2R43 forms an ionic interaction with E318 in the loop between helix α4 and β-strand 6 of the Gαi and Gαgust. S2206.27 of TAS2R43 additionally formed hydrogen bonds with E318 and D341H5.13 of the helices α5 of the Gα subunits.
To investigate the interactions between TAS2Rs and G proteins, we performed a sequence alignment of all TAS2Rs using the residues of TAS2R43 that are involved in interactions with the Gα subunits (Extended Data Fig. 6b,c). I1103.54 and A2276.34 of TAS2R43, which formed hydrophobic interactions with L348H5.20 and L353H5.25 of the Gαi and Gαgust—residues highly conserved in all Gα subunits—are also conserved across TAS2Rs. By contrast, H2125.75, K214ICL3, S2206.27 and H2246.31 of TAS2R43, which formed ionic interactions and hydrogen bonds with E318, D337H5.09 and D341H5.13 of Gαi and Gαgust, are diverse across TAS2Rs (Fig. 3c,e). Residues E318 and D337H5.09 of Gαi and Gαgust are conserved in Gαi/o/z/gust/q/11/14 and Gαi/o/z/gust. Next, we investigated the interactions between the TAS2Rs and Gα subunits in the reported TAS2R14, TAS2R16 and TAS2R46 structures27,28,29,30,31,36 (Supplementary Table 5). D341 of Gαi and Gαgust interacted with H5.68, which is conserved in all TAS2Rs except TAS2R16, where R6.31 instead formed the interaction with D341. D350 of Gαi and Gαgust formed an ionic interaction with K3.53, a conserved residue in all TAS2Rs. Additionally, C351 of Gαi and Gαgust formed hydrophobic interactions with Y3.50 in all of the reported TAS2R structures. These analyses suggest that the interactions might be generally important for Gα binding to TAS2Rs.
To examine the key interactions between TAS2R43 and G proteins, we performed dose–response BRET2 assays on TAS2R43 substitutions, including H205A, K208A, H212A, K214A, S220A and H224A, as well as the double mutants H212A;K214A and H212A;S220A (Fig. 3f–i and Supplementary Tables 6 and 7). H205A and H224A severely impaired activation of both Gi and Ggust. By contrast, other substitutions did not critically affect the receptor activation, while the H212A;K214A double mutant slightly decreased activation that was not statistically significant. Our surface expression measurements showed no statistically significant differences between the mutants and WT. Taken together with our structural data, these results indicate that ionic contacts between TAS2R43 H205, H224 and D341 of the G proteins are critical for G-protein activation.
Structural comparison with other bitter taste receptors
The orthosteric binding pocket engaged by AAI in our TAS2R43 structures is located similar to that in previously reported TAS2R14, TAS2R16 and TAS2R46 structures27,28,36 (Extended Data Fig. 7a–d). The conserved residue W3.32 in each structure typically forms C–π or π–π interactions with ligands, such as cholesterol in the TAS2R14, salicin in the TAS2R16 structure or strychnine in the TAS2R46 structure. This suggests that W3.32 may have a critical role in the recognition of aromatic-ring-containing compounds by bitter taste receptors, including TAS2R43.
Using our TAS2R14 structure28, we performed structural superposition with our TAS2R43 structure (Fig. 4a–c). Notable shifts were observed in the extracellular ends of TM2, TM5, TM6 and TM7 by 6.3 Å (measured between the Cα atoms of V682.63 of TAS2R14 and S682.63 of TAS2R43), 9.3 Å (measured at the Cα atoms of S177ECL2 of TAS2R14 and F174ECL2 of TAS2R43), 5.7 Å (measured at the Cα atoms of V2516.56 of TAS2R14 and V2496.56 of TAS2R43) and 8.8 Å (measured at the Cα atoms of I2627.35 of TAS2R14 and F2617.35 of TAS2R43), respectively (Fig. 4a,b). Additionally, in the aligned structures, TM5 and T863.29 of TAS2R14, which were shifted relative to those of TAS2R43 and substituted for Y853.29 in TAS2R43, respectively, structurally clashed with AAI of our TAS2R43 structure (Fig. 4b). Next, to assess whether AAI could bind within an intracellular region of TAS2R43, we aligned our TAS2R43–Gi structure with the AAI-bound TAS2R14–Gi structure29 (Extended Data Fig. 7e). The alignment revealed a notable displacement of TM6 in TAS2R43 by 3.9 Å (measured at Cα of V6.38) and a repositioning of V6.38 and F6.41 that sterically clashed with AAI in the TAS2R14 structure. F5.58 of TAS2R43 was also rotated, producing an additional clash with AAI. Additionally, the ICL1 and ICL3 were shifted by 7.5 Å (measured between Q39 of TAS2R43 and R39 of TAS2R14) and 6.9 Å (measured between H2125.75 of TAS2R43 and T2155.75 of TAS2R14), respectively (Fig. 4c). In particular, the ordered ICL3 of TAS2R43 was distinct from that of TAS2R14 structure in which the ICL3 and adjacent TM6 segment (G220–T224 of TAS2R14) were partially disordered (Fig. 4c and Extended Data Fig. 7f). The distinct ordered regions, which are extensively involved in interactions with the helix α5 of the Gα subunit including K214ICL3 and S2206.27 of TAS2R43, underlie the distinct G-protein-binding interfaces. Our structural analysis revealed that the interaction between H2055.68 of TAS2R43 and D341 of the helix α5 of the Gα is consistent in the TAS2R14 structure, while H2246.31, involving the interaction with D341, is not conserved in TAS2R14 based on our sequence alignment (Extended Data Figs. 6b,c and 7f). K2085.71, which interacts with D337 of the helix α5 of Gα, is conserved in TAS2R14, while H2125.75 is replaced with T2155.75, which potentially interacts with D337.

a, Structural superposition between the structures of TAS2R43–Gi1 (TAS2R43, orange; Gαiβγ, purple) and TAS2R14–Gi1 (PDB 8VY7; TAS2R14, green; Gαiβγ, blue). Helices are shown as cylinders. b, Extracellular view of a. AAI of TAS2R43 and cholesterol of TAS2R14 are shown as magenta sticks and yellow sticks, respectively. c, Intracellular view of a. G-protein regions except for the α5 helices were omitted for clarity. d, Structural superposition between the structures of TAS2R43–Gi1 (TAS2R43, orange; Gαiβγ, purple) and TAS2R46-mGs/gust (PDB 7XP6; TAS2R46, medium purple; Gαs/gustβγ, pale pink). e, Extracellular view of d. AAI of TAS2R43 and strychnine of TAS2R46 are shown as magenta sticks and cyan sticks, respectively. f, Intracellular view of d. G-protein regions except for the helices α5 were omitted for clarity. g, Structural superposition between the structures of TAS2R43–Gi1 and TAS2R16-Gi1 (PDB 9KPF; TAS2R16, pink; Gαi1βγ, pale green). Structural differences were indicated by black arrows in side view. h, Extracellular view of g. i, Intracellular view of g.
Next, our structural superposition between two structures of TAS2R43 and TAS2R46 (ref. 27) revealed distinct differences, with an r.m.s.d. of 2.5 Å across 275 Cα atoms (Fig. 4d–f). Specifically, the extracellular ends of all 7TM helices in TAS2R46 were shifted from those of TAS2R43, ranging from 2.8 Å (measured at Cα of V2607.43 of TM7) to 6.8 Å (measured at Cα of L712.66 of TM2), unlike the intracellular regions. According to those movements, ECL1 and ECL3 were shifted by 4.9 Å (measured at Cα of L75) and 4.2 Å (measured at Cα of L255) (Fig. 4e). As a result, the TM2, TM4 and TM6 were shifted toward AAI in our TAS2R43 structures compared to the TAS2R46 structure, facilitating hydrophobic interactions by A843.28 and F2526.59, as well as hydrogen bond formation by N652.60, S2486.55 and Y2416.48 (Fig. 4e and Extended Data Fig. 7g). Additionally, Y853.29 was rotated compared to TAS2R43, creating space for AAI binding without a structural clash. Notably, the key residue R2687.42 in TAS2R43, which forms hydrogen bonds and a salt bridge with AAI, is replaced with A2687.42 in TAS2R46.
Recently, salicin-bound TAS2R16 structures were reported36. Superposition of our AAI–TAS2R43–Gi structure with the salicin–TAS2R16–Gi structure revealed clear differences with an r.m.s.d. of 3.8 Å across 259 Cα atoms (Fig. 4g). Overall, the ECLs of TAS2R16 were markedly shifted compared to our TAS2R43 structure (Fig. 4h). In particular, the ECL1 and ECL3 of TAS2R16 moved away from the 7TM core by 5.3 Å (measured between N75 of TAS2R16 and F16 of TAS2R43) and 12.6 Å (measured between L251 of TAS2R16 and G253 of TAS2R43), respectively. Furthermore, TM1, TM3, TM6 and TM7 were displaced by 4.4 Å (measured between F9 of TAS2R16 and I8 of TAS2R43), 4.7 Å (measured between N80 of TAS2R16 and V80 of TAS2R43), 3.0 Å, (measured between T246 of TAS2R16 and S248 of TAS2R43) and 5.8 Å (measured between V260 of TAS2R16 and F263 of TAS2R43), respectively. ICL2 and ICL3 of TAS2R16 were shifted by 8.8 Å (measured between T111 of TAS2R16 and N115 of TAS2R43) and 4.2 Å (measured between N216 of TAS2R16 and G217 of TAS2R43), respectively (Fig. 4i).
To investigate our structures, we aligned our cryo-EM structure of the TAS2R43–Gi complex with a predicted model of the TAS2R43–Gi complex generated using AlphaFold3 (ref. 40). Although the overall structures are similar, showing an r.m.s.d. of 1.1 Å for 281 Cα atoms, a structural superposition revealed that the αN helix of Gαi was shifted and tilted by 8.9 Å and 12.9° (measured at Ala7), respectively (Extended Data Fig. 7h). ECL3 of the predicted model was moved away from the 7TM core by 3.3 Å (measured at S254) resulting in F2526.59 being displaced outward by 1.6 Å in the predicted model. (Extended Data Fig. 7i). Intriguingly, the predicted orthosteric binding pocket in the model was distinct from our structure, with the side chain of Y853.29 rotated, and R2687.42 was slightly shifted toward the AAI molecule, resulting in structural clashes with AAI (Extended Data Fig. 7j). These findings suggest that our cryo-EM structures serve as a more accurate structural template for further elucidating ligand recognition than that provided by AlphaFold3.
Conserved motifs of TAS2R43
The canonical switch motifs, such as D3.49R3.50Y3.51 and N7.49P7.50XXY7.53, for class A GPCR activation are not conserved in class T GPCRs. Instead, different conserved activation-related motifs in class T GPCRs were exhibited in our structures. Specifically, H275, P276 and L279 of the HS/PxxL motif, which is important for TAS2R14 activation as reported, extensively form hydrophobic interactions with A231.49, I271.53, L482.43, F2346.41 and L2376.44 (Extended Data Fig. 7k). This hydrophobic network is conserved in the reported TAS2R46 structure, while the side chain of F2346.41, which is conserved in all TAS2Rs except for TAS2R16, was rotated toward the 7TM core, leading to extensive hydrophobic interactions with A993.43, L1023.46, F1955.58, H2757.49 and Y2717.45 (Extended Data Fig. 7l). Interestingly, water molecule densities were observed near Y1063.50 in both structures of TAS2R43 with either Gi or Ggust (Fig. 3b,d). Our structures revealed that the water molecule forms separate hydrogen bonds with Y1063.50 and the oxygen of the main chain of C351 in Gαi and Gαgust, suggesting that the water-mediated hydrogen bonds may contribute to the interaction between TAS2R43 and Gα subunits.
MD simulation of the TAS2R43 complexes reveals potential cryptic pockets
Our recent study revealed an intracellular site that senses a tastant in the bitter taste receptor TAS2R14. Building upon these findings, we next conducted MD simulations to identify potential cryptic binding pockets within the TAS2R43 structure. First, we carried out five independent simulations of ~0.7 μs for a total of ~3.5 μs of simulation time using the TAS2R43 receptor alone both in the presence and absence of AAI. Additionally, we also carried out three independent simulations of ~0.3 μs for a total of ~0.9 μs of MD simulations using TAS2R43 coupled to the Gi heterotrimer, in both the presence and the absence of AAI. In both simulations, we found that AAI interacting with W883.32 and R2687.42 was stabilized in the orthosteric pocket (Fig. 5a). We then used MDpocket41 to examine any potential cavities that might appear in MD simulations. For the receptor alone, we found notable pockets located at the intracellular side and extending into the core of the receptor. Using just those pockets, we calculated the total pocket volume and found that the apo receptor had overall a larger distribution of pockets than the AAI-bound state (Fig. 5b,c and Supplementary Fig. 1b,c). Additionally, we carried out this analysis on the receptor:heterotrimer simulations and, focusing on just the pockets formed in the receptor core, we found that the pocket volume in the apo state was much more diverse than that of the AAI-bound state (Fig. 5d,e and Supplementary Fig. 1d,e).

a, The r.m.s.d. plots of AAI at the orthosteric pocket of TAS2R43 (left) and the TAS2R43–Gi complex (right), shown as the mean ± s.e.m. across independent MD replicates (n = 5 and n = 3, respectively), respectively. b, Calculated pockets in the AAI-bound TAS2R43 (blue) and apo TAS2R43 (green) structures without G-protein trimers. Black circles indicate pockets at intracellular regions and a purple circle indicates the pocket between the 7TM core and TM4/TM5. c, Representative distribution plots of the total pocket volume in the TAS2R43 structure using the AAI-bound structure or apo model, for cryptic pockets predicted in the simulation. d, Calculated pockets in the AAI-bound TAS2R43 (blue) and apo TAS2R43 (green) structures in complex with Gi trimers. Enlarged views of the calculated intracellular regions are shown in black squares. e, Representative distribution plots of the total pocket volume in the TAS2R43–Gi1 complex, for cryptic pockets predicted in the simulation using the AAI-bound structure or apo model. The total pocket volume (Å3) and their count are indicated on the x axis and y axis, respectively. All of the calculated pockets are shown in each structural model (c,e). f, Predicted movements of the TM4/TM5 in the apo TAS2R43 structure. g, The distance was measured between Cα of V78 in TM3 and Cα of E151 in TM4 in both apo and AAI-bound TAS2R43 structures, either with or without Gi trimers, indicated by the x axis. The y axis indicates the count. Protein Cα r.m.s.d. for all individual simulation runs across all conditions is shown in Supplementary Fig. 1b–e.
While conducting the pocket analysis, we noticed a consistent pocket forming between TM3 and TM4. Examining these simulations further, we decided to investigate the TM dynamics at this spot. We noticed in many of the apo simulations that TM4/TM5 would swing very wide, opening the orthosteric pocket. To investigate this, we used the Cα distance between V78 and E151. Examining the distance distributions in both receptor-only simulations and simulations with the heterotrimer, it is clear that simulations containing AAI favor a more closed pose, whereas, in the apo simulations, these helices open, forming a large pocket for ligands to bind (Fig. 5f,g).
Our MD simulations also suggested that TAS2R43 may possess cryptic pockets within the 7TM domain, allowing the receptor to recognize chemically diverse tastants through multiple binding sites, consistent with previous TAS2R14 structures that revealed intracellular binding regions28. Furthermore, our simulations indicated the potential for constitutive activation of TAS2R43, enabling complex formation with G proteins. On the basis of these observations, we conducted BRET2 assays to evaluate the basal activity of TAS2R43 in the absence of ligands. Our assays revealed that basal activities increased according to increasing transfection amounts of the TAS2R43 gene, showing Gi1 and Ggust dissociation (Extended Data Fig. 8a,b and Supplementary Table 8). The results suggested that TAS2R43 may present highly basal activities, leading to potentially constitutive activation of TAS2R43 to recruit Gi1 and Ggust heterotrimers.
Importantly, our BRET2 assays revealed that AAI activated TAS2R43 for Gi1 and Ggust with higher affinity than coffee compounds and other known tastants, while AAI showed lower Emax than cafestol and kahweol for Ggust signaling (Fig. 2h,i, Extended Data Fig. 8c and Supplementary Tables 4 and 9). The positively charged orthosteric binding pocket appears to be essential for recognizing the acidic compound AAI with relatively higher affinity. To predict the possible binding modes at the orthosteric pocket, we docked the coffee-derived compound cafestol and the known tastant aloin—both of which distinctly activated TAS2R43 in our BRET2 assays (Fig. 2h,i, Extended Data Fig. 8c and Supplementary Tables 4 and 9)—into the orthosteric pocket of our TAS2R43 structure (Extended Data Fig. 9a–c). Because the docking was centered on this region, these models suggest plausible poses at the orthosteric site but do not establish that this pocket is preferred over alternative sites. In this model, the aromatic rings of the compounds were predicted to achieve π–π interactions with W883.32, similar to AAI in our structures. To validate the predicted interactions with W883.32 from docking, we performed dose–response BRET2 assays using WT TAS2R43 and the W88A mutant. The W88A mutant showed remarkedly impaired TAS2R43 activation by AAI and aloin. In contrast, for cafestol, the W88A substitution reduced efficacy by ~50% while increasing affinity compared to WT (Extended Data Fig. 9d–f and Supplementary Table 10).
Furthermore, our MD simulation exhibited instability in TM3 and TM4 but not in the AAI-bound structure. The computational simulation suggests that TM3 and TM4 may be flexible and contribute to variations in the size of the orthosteric pocket, allowing the recognition of ligands of diverse sizes. Ligand binding in the pocket might stabilize TM3 and TM4 simultaneously, in a similar way to AAI in the simulation.
Discussion
Here, we report two cryo-EM structures of AAI-bound TAS2R43–Gi and TAS2R43–Ggust, revealing AAI binding in the orthosteric binding pocket. Our well-resolved structures provided us the opportunity to explore potential water molecules within the 7TM domains, which predicted water-mediated hydrogen bonds between AAI and TAS2R43 residues. This is consistent with our MD simulations, which suggest a potential role for ordered waters in stabilizing AAI binding. Conserved residue W883.32 and selective residue R2687.42 of TAS2R43 could be key residues sensing bitter tastants containing aromatic rings and carboxyl groups with high affinity. These findings are consistent with previous studies on the conserved residue W88 and selective residue R268 (refs. 7,32,39,42). Moreover, F2526.59, which forms a hydrophobic interaction with AAI, may have an even more critical role as a lid for ligand entry than its direct interaction with AAI. This interpretation could be supported by its position at the entrance of the AAI-binding pocket and by the finding that the F2526.59A substitution does not alter AAI-induced efficacy or potency in BRET2 assays but enhances both in cAMP inhibition assays (Fig. 2d–f). Additionally, the structural divergences between TAS2R43 and the reported structures of TAS2R14, TAS2R16 and TAS2R46 suggest that different TAS2R family members broadly recognize chemically diverse tastants. Our functional studies revealed that some coffee-derived chemicals and known bitter tastants activate TAS2R43. Molecular docking positioned a coffee-derived compound cafestol at the orthosteric pocket—consistent with a previous study19—and likewise predicted binding of a known agonist aloin at the same site. Consistent with these predictions, functional assays using the W88A substitution of TAS2R43 affected receptor responses to these agonists. Additionally, MD simulations predicted the presence of potential cryptic allosteric-like pockets, which could provide templates for structure-guided discovery efforts, although further structural studies with diverse agonists and antagonists will be required to understand how these compounds activate or inhibit TAS2R43.
Collectively, our structures of AAI-bound TAS2R43 in complex with Gi and Ggust elucidate the ligand recognition mechanism underlying high-affinity binding. The TAS2R43–Gi and TAS2R43–Ggust complexes demonstrate how TAS2R43 recognizes the toxic compound AAI, initiating signal transduction through both the Gi pathway in extraoral tissues and the Ggust-mediated pathway responsible for bitter taste perception in gustatory nerves, although future structural studies of TAS2R43 for the resolved ECL2, which is functionally unclear, will be required. Overall, our biochemical and computational studies provided a comprehensive analysis of the structures. Taken together, these studies will accelerate the search for selective TAS2R43 modulators as potential therapeutics.
Methods
Constructs
For structure determination, the same genes of Gαi1, Gαgust, Gβ1, Gγ2 and scFv16 were used as previously reported28. A synthesized gene (Integrated DNA Technologies) for human TAS2R43 (UniProt P41159) was cloned into the same modified pFastbac vector as previously described28. Briefly, a fusion cassette comprising an HA signal sequence, a Flag tag, a 10× histidine tag, a tobacco etch virus protease site, thermostabilized apocytochrome BRIL and a 3C (PreScission) protease site was fused at TAS2R43. To enhance the complex formation with G-protein heterotrimers, the NanoBit tethering system was applied for the construct with a fusion of the LgBit at the C terminus of TAS2R43 (ref. 34). The HiBit-fused Gβ1 and the fusion construct of Gγ2 subunit to either dominant negative Gαi1 or Gαgust were cloned into the pFastbac dual vector.
For functional assays, the construct of the HA signal sequence, Flag tag and the N-terminal 45 residues (M1–G45) of rat somatostatin receptor 3 (SST3) were fused to the N terminus of TAS2R43 in pcDNA3.1 vector as previously described28,43. Plasmids encoding the Gαi1, Gαgust, Gβ3, Gγ1 and Gγ9 subunits were obtained from our TRUPATH resources and used according to the TRUPATH protocol38.
All constructs were assembled by PCR using PrimeSTAR polymerase (Takara) and NEBuilder HiFi DNA Assembly (New England Biolabs). All mutants were generated by site-directed mutagenesis (Supplementary Table 11).
Expression and purification of G-protein-coupled TAS2R43 with scFv16
The protein expression and purification of TAS2R43–Gi1–scFv16 and TAS2R43–Ggust–scFv16 were conducted as previously reported28. Briefly, the Bac-to-Bac baculovirus expression system (Invitrogen) was used for generation of TAS2R43, Gαi1, Gαgust, Gβ1, Gγ2 and scFv16 as previously described28. Sf9 cells (Expression Systems ESF921; Mycoplasma free; authenticated by the supplier) at 2.0 × 106 cells per ml were coinfected by baculovirus harboring the TAS2R43 and either Gi1 or Ggust heterotrimers with a multiplicity of infection ratio of 3:1. The cells were further incubated for 48 h at 27 °C. Then, the cells were harvested at 4,000g and cell pellets were stored at −80 °C for future use.
The expression and purification of scFv16 were conducted on the basis of previous studies28,44. Briefly, baculovirus of scFv16 was generated based on Bac-to-Bac system (Invitrogen) with the same protocol as the TAS2R43–G protein complex above. After 4 days of expression at 27 °C, supernatant was harvested by centrifugation and adjusted with 50 mM Tris-HCl (pH 7.5). Chelating reagents were quenched by adding 1 mM NiCl2 and 5 mM CaCl2, followed by stirring at room temperature for 1 h. Supernatant was collected by centrifugation and incubated with His60 Ni Superflow resin (Takara) overnight at 4 °C. On the next day, the resin was washed with 20 column volumes of a washing buffer containing 20 mM HEPES pH 7.5, 500 mM NaCl and 10 mM imidazole. The proteins were eluted with 20 mM HEPES pH 7.5, 100 mM NaCl and 250 mM imidazole. Concentrated proteins were loaded onto a Superdex 200 Increase 10/300 GL column (Cytiva). The main peak fractions containing scFv16 were pooled and concentrated to 2 mg ml−1 and stored at −80 °C for further use.
For purification of the TAS2R43–Gi1–scFv16 and TAS2R43–Ggust–scFv16, the cell pellets expressing each complex were thawed and resuspended in a lysis buffer comprising 20 mM HEPES pH 7.5, 50 mM NaCl, 5 mM MgCl2, 5 mM CaCl2, 50 μM AAI, 300 μM TCEP and protease inhibitors. After incubation for 1 h at room temperature, the pellets were homogenized using a douncer on ice. Then, 50 mU per ml Apyrase (New England Biolabs) and 2 μg ml−1 scFv16 were added and incubated for 30 min at room temperature. After collecting membrane fractions by ultracentrifugation at 72,500g for 30 min (Ti45 rotor, Beckman), the pellets were resuspended in the same lysis buffer in addition with 2 μg ml−1 of scFv16 and 2.5 mU per ml Apyrase (New England Biolabs). Then, a mixture of 0.6% (w/v) lauryl maltose neopentyl glycol (LMNG; Anatrace) and 0.12% (w/v) cholesteryl hemisuccinate tris salt (CHS; Anatrace) was added to solubilize the complex proteins and incubated for 3 h at 4 °C. After removing insoluble fractions by ultracentrifugation at 142,000g for 30 min (Ti45 rotor, Beckman), supernatants in addition with 20 mM imidazole were incubated with a TALON IMAC resin (Takara) for overnight at 4 °C. The resin was washed with 20 column volumes of washing buffer comprising 20 mM HEPES pH 7.5, 100 mM NaCl, 1 mM MgCl2, 0.01% LMNG, 0.001% CHS, 30 mM imidazole and 50 μM AAI using an Econo-Pac gravity column (Bio-Rad). The proteins were eluted in a buffer comprising 20 mM HEPES pH 7.5, 100 mM NaCl, 1 mM MgCl2, 0.01% LMNG, 0.001% CHS, 300 mM imidazole and 50 μM AAI. The eluants were incubated with 0.2 mg of scFv16 for 30 min at 4 °C and then loaded onto a Superose 6 10/300 GL column (Cytiva) in a running buffer containing 20 mM HEPES pH 7.5, 100 mM NaCl, 0.00075% LMNG, 0.00015% CHS, 0.00025% GDN, 300 μM TCEP and 50 μM AAI. Peak fractions containing the complex were pooled, concentrated and subsequently loaded onto a Superose 6 10/300 GL column (Cytiva) equilibrated with the same running buffer. The pooled proteins from homogeneous main peak fractions were concentrated to 2–3 mg ml−1 using an Amicon Ultra centrifugal filter (100-kDa cutoff; Millipore).
Cryo-EM data collection and processing
Cryo-EM sample preparation and data collection were conducted as previously described28. Briefly, 3.3 μl of proteins for TAS2R43–Gi–scFv16 and TAS2R43–Ggust–scFv16 were blotted onto glow-discharged holey carbon grids (Quantifoil R1.2/1.3 300-mesh, Ted Pella). The grids were plunge-frozen into a liquid ethane–propane (40:60) mixture using a Vitrobot Mark IV (Thermo Fisher Scientific) after blotting with a blot force of −10 for 4 s at 4 °C and 100% humidity. Data were collected using a 200-keV Talos Arctica equipped with a Gatan K3 direct electron detector with a nominal magnification of ×45,000, corresponding to a pixel size of 0.876 Å. Automatic image acquisition was performed using SerialEM with an exposure dose rate of ~15–17 electrons per pixel per second in counting mode. Images were recorded for ~3 s in 60 subframes corresponding to ~55–67 electrons per Å2.
For the dataset of the TAS2R43–Gi–scFv16 complex, data processing was performed using cryoSPARC (version 4.2)45. After patch-based motion correction and contrast transfer function (CTF) estimation, particles were initially picked using blob picking. The extracted particles underwent three rounds of two-dimensional (2D) classification to remove junk particles. Selected particles from the final 2D averages were used for three-dimensional (3D) classification, followed by nonuniform (NU) refinement, yielding a 3.12-Å reconstruction. In parallel, the good particles from the final 2D averages were used to generate templates for template-based particle picking. The newly picked particles were extracted and subjected to three rounds of 2D classification, followed by 3D classification. Particles from the best 3D classes were then refined using NU refinement, yielding a 3.13-Å map. Particle sets from the 3.12-Å and 3.13-Å reconstructions were combined and duplicates were removed. The combined particles were used for Topaz particle picking and the picked particles were subjected to 2D classification to exclude junk particles, followed by another round of duplicate removal. These particles were then combined again with the previously obtained 3.12-Å and 3.13-Å particle sets, followed by final duplicate removal. Subsequent 3D classification was performed and the best classes were selected for NU refinement. Global CTF refinement and local motion correction were then applied, resulting in a final globally refined map at 2.91-Å resolution. Lastly, local refinement was conducted using masks for the TAS2R43 receptor and the Gαi1β1γ2–scFv16 complex, yielding locally refined maps at 3.10-Å and 2.81-Å resolution, respectively. Negative B factors determined automatically in cryoSPARC (version 4.2)45 (−151.6 and −114.4 for TAS2R43 and Gαi1β1γ2–scFv16 local maps, respectively) were applied for map sharpening.
For the dataset of the TAS2R43–Ggust–scFv16 complex, data processing was conducted using cryoSPARC (version 4.2)45. Following patch-based motion correction and CTF estimation, initial particle picking was performed using blob picking. Extracted particles were subjected to three rounds of 2D classification to discard junk particles. Selected particles from the final 2D averages proceeded through two rounds of 3D classification, followed by NU refinement, resulting in a 3.24-Å reconstruction. These refined particles were then used to generate templates for a second round of template-based particle picking. The newly picked particles underwent three additional rounds of 2D classification and subsequent 3D classification. The best-resolved 3D classes were refined using NU refinement, again producing a 3.24-Å map. Particle sets from both 3.24-Å reconstructions were merged and used to train the Topaz particle picker. Topaz-selected particles were extracted and further refined through 2D classification and poor-quality particles were excluded. After duplicate removal, the refined particles were subjected to 3D classification and high-quality classes were selected. These particles were then combined with the earlier two 3.24-Å datasets, followed by another round of duplicate removal. Additional 3D classification and NU refinement yielded a 3.11-Å reconstruction. Global CTF refinement and local motion correction were subsequently applied, producing a final globally refined map at 3.04-Å resolution. Lastly, focused local refinements were carried out using masks targeting the TAS2R43 receptor and the Gαgustβ1γ2–scFv16 complex, resulting in locally refined maps at resolutions of 3.21 Å and 2.88 Å, respectively. Negative B factors determined automatically in cryoSPARC (version 4.2)45 (−146.8 and −116.1 for TAS2R43 and Gαgustβ1γ2–scFv16 local maps, respectively) were applied for map sharpening.
Model building and refinement
Composite maps generated using ChimeraX46 were used for model building and refinement. The prediction model for TAS2R14 generated by AlphaFold3 (ref. 40) was used as an initial model for GPCR regions. Previously reported structures for the TAS2R14–Gαi1β1γ2–scFv16 (PDB 8VY7) and TAS2R14–Gαgustβ1γ2–scFv16 (PDB 8VY9) were used as initial templates for the corresponding G protein–scFv16 regions28. Further model adjustment and real-space refinement were performed in in Coot (version 0.9.8)47 and PHENIX48. Model quality was assessed with the comprehensive cryo-EM validation tools in PHENIX and ligand placement was evaluated using GemSpot35. Water molecules were built using phenix.douse in PHENIX48. The model statistics were assessed using MolProbity49. All structural figures were prepared in ChimeraX46.
BRET2 assays
BRET2 assays were conducted in accordance with the published TRUPATH protocol and its associated resources38. For dose–response BRET2 assays, 0.1 million HEK293T cells (American Type Culture Collection (ATCC), CRL-11268; Mycoplasma free; authenticated by the supplier) were seeded in six-well plates 24 h before transfection. Plasmid DNAs (500 ng of SST3–TAS2R43, 200 ng of Gαi1–Rluc or Gαgust–Rluc, 100 ng of Gβ3 and 100 ng of GFP2–Gγ9 or GFP2–Gγ1) were mixed with TransIT-2020 (MirusBio) in Opti-MEM (Gibco) and added to HEK293T cells cultured in DMEM containing 10% FBS, 1 IU per ml penicillin G and 100 μg ml−1 streptomycin. After 24 h, transfected cells were washed with PBS, trypsinized and then plated at 10,000 cells per well onto poly(L-lysine)-coated 384-well white, clear-bottom plates (Greiner) in DMEM containing 1% dialyzed FBS, 1 IU per ml penicillin G and 100 μg ml−1 streptomycin. After 12–18 h, the cells were washed with a drug buffer containing 1× Hanks’ balanced salt solution (HBSS), 20 mM HEPES pH 7.4, 0.1% (w/v) BSA and 0.01% (w/v) ascorbic acid. Drugs prepared in drug buffer were applied to the cells and coelenterazine 400a (Nanolight) was then dispensed immediately to a final concentration of 5 μM. Plates were incubated for 15 min at room temperature before measurement. BRET2 signals were acquired on a PHERAstar plate reader by recording Rluc emission at 400 nm and GFP2 fluorescence emission at 515 nm with a 1-s integration time per well. The BRET2 ratio (GFP2/Rluc) was calculated for each well and concentration–response curves were generated in GraphPad Prism (version 10.4.1).
Basal activity was assessed using the same workflow as above, except that cells were transfected with plasmids at a receptor:Gα–Rluc:Gβ:GFP2–Gγ DNA ratio of 5:0.25:1:1.
Split luciferase biosensor cAMP inhibition assay
cAMP inhibition assays were conducted as previously reported28. Briefly, HEK293T cells (ATCC, CRL-11268; Mycoplasma free; authenticated by the supplier) were plated at 2.5 × 106 cells per 10-cm dish 24 h before transfection. On the following day, cells were transiently transfected with 5 μg of TAS2R43 plasmid and 1 μg of the cAMP biosensor plasmid (GloSensor 22F; Promega) in DMEM supplemented with 10% (v/v) FBS, 1 IU per ml penicillin G and 100 μg ml−1 streptomycin. Then, 24 h after transfection, cells were detached and plated onto poly(L-lysine)-coated 384-well clear-bottom plates (Greiner) at 10,000 cells per well in DMEM containing 1% dialyzed FBS, 1 IU per ml penicillin G and 100 μg ml−1 streptomycin. After an additional 24 h, culture medium was aspirated and wells were washed with 20 μl per well of assay (drug) buffer (1× HBSS, 20 mM HEPES pH 7.4, 0.01% (w/v) ascorbic acid and 0.1% (w/v) BSA). Fresh assay buffer (20 μl per well) was then added, followed by addition of 10 μl of 3× drug solutions prepared in the same buffer and incubation for 15 min at room temperature. Next, 10 μl of drug buffer containing luciferin and isoproterenol was added to yield final concentrations of 1 mM and 200 nM, respectively, followed by incubation for 15 min at room temperature. Luminescence was measured and data were processed and plotted using GraphPad Prism (version 10.4.1).
MD simulations
In this work, we carried out a total of four simulation sets: (1) TAS2R43 bound to AAI and the Gi heterotrimer; (2) apo TAS2R43 with the Gi heterotrimer; (3) TAS2R43 bound to AAI; and (4) apo TAS2R43.
For MD simulations of the receptor–heterotrimer (Gi) complexes (simulations 1 and 2), the scFv16 was removed from the coordinate files. The missing portions of the Gα subunit N terminus and the Gγ subunit C terminus were reconstructed and modified with appropriate lipid attachments50. Specifically, myristoylation was applied to G2 of the Gα subunit, palmitoylation to C3 of the Gα subunit and prenylation to C68 of the Gγ subunit, in accordance with established lipidation patterns. Any unresolved residues in the Gα subunit were incorporated into the model using Maestro’s ‘crosslink’ function. The receptor’s missing residues were modeled using coordinates generated from the AlphaFold3 (ref. 40) prediction. In the apo receptor simulations, the coordinates for AAI were removed. In simulations of the receptor alone, the heterotrimer and scFv16 were removed from the coordinates as appropriate. Identical simulation procedures were followed for all systems thereafter.
Each coordinate set underwent preparation using Maestro’s protein preparation wizard (version 13.3; Schrodinger), followed by alignment to a previous structure (PDB 7XP6)27 according to the Orientation of Protein in Membranes database51. The CHARMM-GUI membrane bilayer builder was then used to build the simulations52,53,54,55. The receptor or receptor–heterotrimer complexes were embedded in a palmitoyl oleoyl phosphatidylcholine bilayer with 150 mM sodium chloride for neutralization. CHARMM-GUI generated parameters for AAI during system construction.
Five independent simulations lasting approximately 1.4 μs each were conducted with randomized initial velocities for the receptor-only simulations, resulting in a total of 7 μs of simulation time per system. The heterotrimer-containing simulations comprised three independent runs of approximately 380 ns each with randomized velocities, totaling about 1.14 μs of simulation time.
For the TAS2R43–AAI complex, two simulation sets—one including and one excluding the resolved waters—were performed to assess their effect. In the water-included condition, four independent trajectories (0.5–1 μs each) were initiated with randomized velocities. Following CHARMM-GUI’s standard minimization and six-step equilibration protocol (totaling 1.875 ns; 125 ps × 3 + 500 ps × 3), during which positional and dihedral restraints on the protein and lipids were gradually reduced. They followed the same production protocol described below.
All simulations used the CHARMM36m56 force field and the TIP3P57 water model using AMBER20 (ref. 58) with a CUDA-accelerated version of particle mesh Ewald MD. Following CHARMM-GUI’s recommended minimization and equilibration protocols, production runs were executed in an NPT ensemble (310 K, 1 bar) using a Langevin thermostat59 and Monte Carlo barostat60 with a 2.0-fs time step. Electrostatic interactions were calculated using the particle mesh Ewald method61 with nonbonded interaction cutoffs at 9.0 Å. The SHAKE62 algorithm constrained hydrogen bond lengths.
Pytraj was used for trajectory alignment and reimaging, while r.m.s.d. calculations were performed in MDAnalysis63,64. MDpocket was used to calculate all pockets during simulations41. ChimeraX and VMD were used for the molecular visualizations, while other plots were created using seaborn and matplotlib in Python46,65.
Molecular docking
AAI, cafestol and aloin were docked to the orthosteric binding pocket in our Gi-coupled TAS2R43 structure using Glide (Schrodinger). Briefly, the receptor was prepared in Maestro (version 13.9; Schrodinger). Ligands were prepared using LigPrep (Schrodinger). The docking grid center was determined on the basis of the position of AAI in the structure. Extra precision docking was conducted in Glide and the top scored binding pose from five poses per ligand was visualized using ChimeraX46.
Cell surface expression of receptors
Cell surface expressions of WT and mutant TAS2R43 were measured using ELISA chemiluminescence, as previously reported66. In brief, after 48 h of transfection, cells grown in white 384-well plates were fixed by adding 20 μl of 4% (v/v) paraformaldehyde per well and incubating for 10 min at room temperature. The cells were washed twice with 40 μl of PBS, followed by blocking with 20 μl of 5% (v/v) BSA in PBS for 1 h. Cells were then incubated for 1 h at room temperature with an anti-Flag antibody conjugated to horseradish peroxidase (Sigma-Aldrich, A8592) diluted 1:5,000 in 5% (v/v) BSA in PBS. After three washes with 80 μl of PBS, 20 μl of SuperSignal ELISA Pico substrate (Thermo Fisher, 37070) was added to each well for signal detection. Luminescence was measured using a PHERAstar FSX plate reader (BMG LABTECH). Data analysis was performed in GraphPad Prism (version 10.6) and signals were normalized to those from WT TAS2R43.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Atomic coordinates and cryo-EM maps were deposited to the Protein Data Bank and EM Data Bank under the accession codes PDB 9OXA, EMD-70964 and PDB 9OXB, EMD-70965 for the TAS2R43–Gi1–scFv16 and TAS2R43–Ggust–scFv16 complexes, respectively. Simulation trajectories and ligand parameters generated in this study are available from Zenodo (https://doi.org/10.5281/zenodo.18404816)67. Additional data and materials are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Chandrashekar, J., Hoon, M. A., Ryba, N. J. & Zuker, C. S. The receptors and cells for mammalian taste. Nature 444, 288–294 (2006).
Mueller, K. L. et al. The receptors and coding logic for bitter taste. Nature 434, 225–229 (2005).
Zhao, G. Q. et al. The receptors for mammalian sweet and umami taste. Cell 115, 255–266 (2003).
Zhang, Y. et al. Coding of sweet, bitter, and umami tastes: different receptor cells sharing similar signaling pathways. Cell 112, 293–301 (2003).
Roper, S. D. & Chaudhari, N. Taste buds: cells, signals and synapses. Nat. Rev. Neurosci. 18, 485–497 (2017).
Chun, L., Zhang, W. H. & Liu, J. F. Structure and ligand recognition of class C GPCRs. Acta Pharmacol. Sin. 33, 312–323 (2012).
Di Pizio, A. et al. Comparing class A GPCRs to bitter taste receptors: structural motifs, ligand interactions and agonist-to-antagonist ratios. Methods Cell. Biol. 132, 401–427 (2016).
Jin, H., Fishman, Z. H., Ye, M., Wang, L. & Zuker, C. S. Top-down control of sweet and bitter taste in the mammalian brain. Cell 184, 257–271 (2021).
Tuzim, K. & Korolczuk, A. An update on extra-oral bitter taste receptors. J. Transl. Med. 19, 440 (2021).
Ziaikin, E., David, M., Uspenskaya, S. & Niv, M. Y. BitterDB: 2024 update on bitter ligands and taste receptors. Nucleic Acids Res. 53, D1645–D1650 (2025).
Suess, B., Brockhoff, A., Meyerhof, W. & Hofmann, T. The odorant (R)-citronellal attenuates caffeine bitterness by inhibiting the bitter receptors TAS2R43 and TAS2R46. J. Agric. Food Chem. 66, 2301–2311 (2018).
Pirastu, N. et al. Association analysis of bitter receptor genes in five isolated populations identifies a significant correlation between TAS2R43 variants and coffee liking. PLoS ONE 9, e92065 (2014).
Wang, Q., Liszt, K. I. & Depoortere, I. Extra-oral bitter taste receptors: new targets against obesity? Peptides 127, 170284 (2020).
Kim, K. S., Egan, J. M. & Jang, H. J. Denatonium induces secretion of glucagon-like peptide-1 through activation of bitter taste receptor pathways. Diabetologia 57, 2117–2125 (2014).
Grassin-Delyle, S. et al. The expression and relaxant effect of bitter taste receptors in human bronchi. Respir. Res. 14, 134 (2013).
Shah, A. S., Ben-Shahar, Y., Moninger, T. O., Kline, J. N. & Welsh, M. J. Motile cilia of human airway epithelia are chemosensory. Science 325, 1131–1134 (2009).
Liszt, K. I. et al. Human intestinal bitter taste receptors regulate innate immune responses and metabolic regulators in obesity. J. Clin. Invest. 132, e144828 (2022).
Meyerhof, W. et al. The molecular receptive ranges of human TAS2R bitter taste receptors. Chem. Senses 35, 157–170 (2010).
Lang, T. et al. Numerous compounds orchestrate coffee’s bitterness. J. Agric. Food Chem. 68, 6692–6700 (2020).
Czech, C. et al. Identification of mozambioside roasting products and their bitter taste receptor activation. Food Chem. 446, 138884 (2024).
Konstantinidis, N., Franke, H., Schwarz, S. & Lachenmeier, D. W. Risk assessment of trigonelline in coffee and coffee by-products. Molecules 28, 3460 (2023).
Lemos, M. F. et al. Chemical and sensory profile of new genotypes of Brazilian Coffea canephora. Food Chem. 310, 125850 (2020).
Farah, A., de Paulis, T., Trugo, L. C. & Martin, P. R. Effect of roasting on the formation of chlorogenic acid lactones in coffee. J. Agric. Food Chem. 53, 1505–1513 (2005).
del Castillo, M. D., Ames, J. M. & Gordon, M. H. Effect of roasting on the antioxidant activity of coffee brews. J. Agric. Food Chem. 50, 3698–3703 (2002).
Muller, C., Hemmersbach, S., Slot, G. V. & Hofmann, T. Synthesis and structure determination of covalent conjugates formed from the sulfury-roasty-smelling 2-furfurylthiol and di- or trihydroxybenzenes and their identification in coffee brew. J. Agric. Food Chem. 54, 10076–10085 (2006).
Ginz, M. & Engelhardt, U. H. Identification of proline-based diketopiperazines in roasted coffee. J. Agric. Food Chem. 48, 3528–3532 (2000).
Xu, W. et al. Structural basis for strychnine activation of human bitter taste receptor TAS2R46. Science 377, 1298–1304 (2022).
Kim, Y. et al. Bitter taste receptor activation by cholesterol and an intracellular tastant. Nature 628, 664–671 (2024).
Hu, X. et al. Bitter taste TAS2R14 activation by intracellular tastants and cholesterol. Nature 631, 459–466 (2024).
Tao, L. et al. Bitter taste receptor TAS2R14 activation and G protein assembly by an intracellular agonist. Cell Res. 34, 735–738 (2024).
Peri, L. et al. A bitter anti-inflammatory drug binds at two distinct sites of a human bitter taste GPCR. Nat. Commun. 15, 9991 (2024).
Brockhoff, A., Behrens, M., Niv, M. Y. & Meyerhof, W. Structural requirements of bitter taste receptor activation. Proc. Natl Acad. Sci. USA 107, 11110–11115 (2010).
Lang, R., Lang, T., Dunkel, A., Ziegler, F. & Behrens, M. Overlapping activation pattern of bitter taste receptors affect sensory adaptation and food perception. Front. Nutr. 9, 1082698 (2022).
Inoue, A. et al. Illuminating G-protein-coupling selectivity of GPCRs. Cell 177, 1933–1947 (2019).
Robertson, M. J., van Zundert, G. C. P., Borrelli, K. & Skiniotis, G. GemSpot: a pipeline for robust modeling of ligands into cryo-EM maps. Structure 28, 707–716 (2020).
Wang, X. et al. Structural basis of β-glucopyranoside salicin recognition by a human bitter taste GPCR. Cell Rep. 44, 115604 (2025).
Shub, L., Liu, W., Skiniotis, G., Keiser, M. J. & Robertson, M. J. MIC: a deep learning tool for assigning ions and waters in cryo-EM and crystal structures. Nat. Commun. 16, 6182 (2025).
Olsen, R. H. J. et al. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol. 16, 841–849 (2020).
Sandal, M. et al. Evidence for a transient additional ligand binding site in the TAS2R46 bitter taste receptor. J. Chem. Theory Comput. 11, 4439–4449 (2015).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Schmidtke, P., Bidon-Chanal, A., Luque, F. J. & Barril, X. MDpocket: open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics 27, 3276–3285 (2011).
Pronin, A. N., Tang, H., Connor, J. & Keung, W. Identification of ligands for two human bitter T2R receptors. Chem. Senses 29, 583–593 (2004).
Bufe, B., Hofmann, T., Krautwurst, D., Raguse, J. D. & Meyerhof, W. The human TAS2R16 receptor mediates bitter taste in response to β-glucopyranosides. Nat. Genet. 32, 397–401 (2002).
Koehl, A. et al. Structure of the micro-opioid receptor–Gi protein complex. Nature 558, 547–552 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Wedegaertner, P. B., Wilson, P. T. & Bourne, H. R. Lipid modifications of trimeric G proteins. J. Biol. Chem. 270, 503–506 (1995).
Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I. & Lomize, A. L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 (2012).
Brooks, B. R. et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 (2009).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Lee, J. et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 12, 405–413 (2016).
Wu, E. L. et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 35, 1997–2004 (2014).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Mark, P. & Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 105, 9954–9960 (2001).
Case, D. A. et al. Recent developments in Amber biomolecular simulations. J. Chem. Inf. Model. 65, 7835–7843 (2025).
Loncharich, R. J., Brooks, B. R. & Pastor, R. W. Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers 32, 523–535 (1992).
Åqvist, J. et al. Molecular dynamics simulations of water and biomolecules with a Monte Carlo constant pressure algorithm. Chem. Phys. Lett. 384, 288–294 (2004).
Salomon-Ferrer, R. et al. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 9, 3878–3888 (2013).
Ryckaert, J. P., Ciccotti, G. & Berendsen, H. J. C. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
Roe, D. R. & Cheatham, T. E. III. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 (2013).
Michaud-Agrawal, N., Denning, E. J., Woolf, T. B. & Beckstein, O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32, 2319–2327 (2011).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Zhang, S. et al. Inactive and active state structures template selective tools for the human 5-HT5A receptor. Nat. Struct. Mol. Biol. 29, 677–687 (2022).
Kim, Y. et al. Extended data for the publication: Structural insights into coffee bitter taste perception by TAS2R43 receptor. Zenodo https://doi.org/10.5281/zenodo.18404816 (2026).
Acknowledgements
This work was supported by the National Institute of Mental Health Psychoactive Drug Screening Program (U24DK116195), the Michael Hooker Distinguished Professorship to B.L.R. and National Institutes of Health grant R35GM158122 to R.O.D. We acknowledge E. Robinson, C. Lenger and J. Strauss of the University of North Carolina cryo-EM core facility, supported by the National Cancer Institute of the National Institutes of Health under award number P30CA016086, for technical assistance in this project.
Author information
Authors and Affiliations
Contributions
Y.K. and B.L.R. designed the project. Y.K. performed the cloning and the protein expression and purification, prepared the cryo‑EM grids, processed the cryo‑EM data, carried out the model building and refinement, analyzed the structural data, conducted the molecular docking, cAMP inhibition assays and BRET assays, wrote the manuscript and prepared all the figures. R.H.G. performed the MD simulations and prepared the relevant figures. R.H.G. edited the manuscript. Y.Z. performed and analyzed the MD simulations, with guidance from R.O.D. B.L.R. supervised the entire project and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
B.L.R. is a cofounder of ImprintBio, Lassogen and Epiodyne and a scientific advisory board member of Septerna and Lassogen. The University of North Carolina has licensed TRUPATH technology to many pharmaceutical and biotechnology companies. R.O.D. is a scientific advisor to Septerna. The other authors declare no competing interests.
Peer review
Peer review information
Nature Structural & Molecular Biology thanks Fei Xu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Katarzyna Ciazynska, in collaboration with the Nature Structural & Molecular Biology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Cryo-EM data processing for TAS2R43–Gi1–scFv16 complex.
a, Workflow of cryo-EM data processing for the TAS2R43–Gi1–scFv16 complex. b, Representative motion-corrected micrograph randomly selected out of 2,253 similar micrographs at a nominal × 81,000 magnification (left) and 2D averages (right) for TAS2R43–Gi1–scFv16 complex. c, Particle angular distributions for a globally refined map for TAS2R43–Gi1–scFv16 complex. d, Gold-Standard Fourier shell correlation (GSFSC) curves of locally refined maps for the TAS2R43 (left) and Gi-scFv16 (right).
Extended Data Fig. 2 Cryo-EM data processing for TAS2R43–Ggust–scFv16 complex.
a, Workflow of cryo-EM data processing for the TAS2R43–Ggust–scFv16 complex. b, Representative motion-corrected micrograph randomly selected out of 4,175 similar micrographs at a nominal × 81,000 magnification (left) and 2D averages (right) for TAS2R43–Ggust–scFv16 complex. c, Particle angular distributions for a globally refined map for TAS2R43–Ggust–scFv16 complex. d, Gold-Standard Fourier shell correlation (GSFSC) curves of locally refined maps for the TAS2R43 (left) and Ggust–scFv16 (right).
Extended Data Fig. 3 Cryo-EM density and model for TAS2R43–Gi–scFv16 and TAS2R43–Ggust–scFv16.
Representative cryo-EM density and model for the TM helices, ICLs, ECLs and AAI of TAS2R43 and the helix α5 of the Gα subunit of for TAS2R43–Gi1–scFv16 (a) and TAS2R43–Ggust–scFv16 (b).
Extended Data Fig. 4 Structural features of TAS2R43 structures.
a,b, Modeled AAI densities were validated using Gemspot software. c,d, Distinct ICL2 helices, conserved in TAS2R43 (orange), TAS2R14 (PDB 8VY7; green), and TAS2R46 (PDB 7XP6) (c), extend TM5 of TAS2R43 compared to TAS2R14 and TAS2R46 (d). e, Orthosteric binding pocket occupied by AAI and F252 was shown as a green circle. f, Structural superposition between the structures of TAS2R43–Gi1 (TAS2R43, orange; Gαi1, purple; Gβ1, sky blue) and TAS2R43-Ggust (TAS2R43, brown; Gαgust, pink; Gβ1, blue) complexes, highlighting the proximity of TAS2R43 ICL1 and ICL2 to Gβ1 and to the αN helices in both Gα subunits. A black square indicated a potential interaction between the ICL1 of TAS2R43 and Gβ1. g, Enlarged view of a potential interaction between R38 in the ICL1 and D312 of Gβ1 for the TAS2R43–Gi1 structure. h, Same view for the TAS2R43-Ggust complex as in g.
Extended Data Fig. 5 Structural details of water molecules in the structures of TAS2R43–Gi1 and TAS2R43–Ggust.
a, Representative structural superposition of TAS2R43–Gi1 (orange) and TAS2R43–Ggust (green). b,c, Representative structural models for the TAS2R43–Gi1 (b) and TAS2R43–Ggust (c). The water molecules and their EM densities are shown as red spheres and green meshes, respectively. Enlarged views of the orthosteric binding pocket of the TAS2R43 highlighted AAI, water molecules and interacting residues (a-c). The water molecules were indicated as red spheres with EM densities shown as black meshes. Black dashed lines indicate hydrogen bonds. Water molecules in the interfaces between TAS2R43 and either Gi1 or Ggust were shown in Fig. 3b, d. d-f, Previously reported cryo-EM maps and structural models showing potential water densities at the interfaces between TAS2Rs and Gα subunits. The potential water densities, indicated by red dashed circles, were observed in the structures of the TAS2R14–Gi1 (d) and TAS2R14–Ggust (e,f). TAS2R14 models are shown in green or blue, and the Gα subunits are shown in purple or pink. g-k, MD simulations of TAS2R43–Gi1 structures under three conditions: AAI-bound with resolved waters (AAI+water), AAI-bound without resolved waters, and apo with resolved waters. g, Average predicted water density (green mesh) overlaid on the AAI-bound cryo-EM structure; built waters, red spheres. h, Number of waters at the W4 site (waters hydrogen bonding to both W88 and Y241), mean ± s.e.m.; dots indicate n = 4–5 independent replicates. i, In AAI-bound simulations, W4 remains occupied; in apo simulations, the site shifts with pocket rearrangement (gray→blue) but remains hydrated. j, Ligand RMSD relative to the resolved pose: omission of W4 water destabilizes AAI binding. k, Water density from MD simulations. In the AAI-bound TAS2R43 simulations with resolved water molecules, the W4 water site remains consistently occupied, and AAI density matches the resolved AAI placement. In simulations without resolved water, AAI shifts away from its resolved binding mode. Protein Cα RMSDs for the individual runs across the simulations for water placement are shown in Supplementary Fig. 1a.
Extended Data Fig. 6 Sequence alignment of interacting residues of TAS2R43 and Gαi1/gust.
a, Sequences of TAS2Rs were aligned based on the AAI-interacting residues in the orthosteric binding pocket of TAS2R43. b, Sequences of TAS2Rs were aligned based on the residues of TAS2R43 involved in the G protein-binding interface. c, Sequences of all Gα subunits were aligned based on the residues of the Gαi1 and Gαgust that interact with TAS2R43. The alignment was conducted based on GPCRdb (https://gpcrdb.org).
Extended Data Fig. 7 Structure comparison of TAS2R43 with other taste receptors.
a-d, Representative structures for AAI-bound TAS2R43 coupled with Gi (orange) (a), cholesterol-bound TAS2R14 coupled with Gi (green, PDB 8VY7) (b), strychnine-bound TAS2R46-miniGs/gust (medium purple, PDB 7XP6) (c), and salicin-bound TAS2R16-Gi1 (pink, PDB 9KPF) (d). AAI, cholesterol and strychnine were indicated by spheres and colored magenta, yellow, and cyan, respectively. Dashed circles indicated W88(89)3.32 and a dashed line indicated the relative position of the residue in the structures. e, Structural clashed between our TAS2R43-Gi structure (orange) and AAI in TAS2R14-Gi1 (green, PDB 8XQO). Hydrogen bonds were indicated by black dashed lines. f, Interacting residues (sticks) in the interface between GPCR and the Gαi in the TAS2R43-Gi (TAS2R43, orange; Gi, purple) and TAS2R14-Gi (TAS2R14, green; Gi, blue). A black dashed circle indicates disordered regions of TAS2R14, resulting in structural differences. g, Structural superposition of the TAS2R43-Gi (orange) and TAS2R46-mGs/gust (PDB 7XP6, medium purple) highlighting the residues in the orthosteic binding pocket. Strychnine of the TAS2R46 structure was omitted for clarity. h-k, Structural superposition of the cryo-EM structure of TAS2R43-Gi (TAS2R43, orange; G proteins, purple) and the AlphaFold3-predicted model of TAS2R43-Gi (TAS2R43, medium blue; G proteins, cyan). The superposition, shown in an extracellular view, highlights structural differences in the αN helix of Gα (h) and in the ECL3 (i). An enlarged view of the orthosteric pocket is shown. Black dashed circles indicate structural clashes between the AlphaFold3 model and the AAI ligand observed in the cryo-EM structure. k,l, Interactions of the HPxxL motif with surrounding residues were highlighted.
Extended Data Fig. 8 Basal activities and surface expression levels of TAS2R43.
a,b, Basal activities of TAS2R43 coupled with Gi1 (a) and Ggust (b) measured by BRET2 assays using HEK293T cells. The total amounts of transfected DNA were fixed using pcDNA3.1. Data represent mean ± s.e.m. of n = 3 biological replicates. c, An agonist-stimulated dose-response BRET2 assay for TAS2R43-Gi1. Data represent mean ± s.e.m. of n = 4 biological replicates. d, Surface expression of all TAS2R43 mutants used in this study was measured by ELISA in HEK293T cells. The values were normalized to WT based on pcDNA3.1-transfected cells. Data represent mean ± s.e.m. of n = 3 biological replicates. Significant differences were determined by ordinary one-way ANOVA with Dunnett’s test for multiple comparisons based on the values of pcDNA3.1. Exact P values were indicated. Statistical significance was denoted as ****P < 0.0001, ***P < 0.001, **P < 0.01, and *P < 0.05, and ns (not significantly different) (a,b,d). All of the ΔBRET2, pEC50 and Emax values for functional assays are provided in Supplementary Tables 7 and 8 (a-c).
Extended Data Fig. 9 Molecular docking of cafestol and aloin for TAS2R43.
a-c, Representative docked AAI (a), cafestol (b), and aloin (c) into the TAS2R43–Gi1 structure (top) together with their chemical structures (middle) and agonist-stimulated BRET2 assays using W88A mutant (bottom). All docked ligands are shown as stick models. The docked AAI exhibited the same binding pose as observed in the TAS2R43–Gi1 structure. Docked AAI, cryo-EM map-based-modeled AAI based on cryo-EM map, docked cafestol, and docked aloin are shown as magenta, cyan, orange, and pink sticks, respectively. The docking scores (XP GlideScore) obtained from XP docking were −11.3, −7.2, and −11.1 kcal mol−1 for AAI, cafestol and aloin, respectively. d-f, Agonist-stimulated dose-response BRET2 assays for Gi1 using TAS2R43 W88A mutants at the orthosteric pocket with aristolochic acid I (d), cafestol (e), and aloin (f). Values were normalized to WT. Data represent mean ± s.e.m. of n = 3 biological replicates.
Supplementary information
Supplementary Information (download PDF )
Supplementary Fig. 1 and Tables 1–11.
Source data
Source Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 8 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 9 (download XLSX )
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, Y., Gumpper, R.H., Zhuang, Y. et al. Structural insights into coffee bitter taste perception by TAS2R43 receptor. Nat Struct Mol Biol 33, 701–710 (2026). https://doi.org/10.1038/s41594-026-01776-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41594-026-01776-w
