
Abstract
Nervonic acid (C24:1 Δ15, NA) is a very long-chain monounsaturated fatty acid, a clinically indispensable resource in maintaining the brain and nerve cells development and regeneration. Till now, NA has been found in 38 plant species, among which the garlic-fruit tree (Malania oleifera) has been evaluated to be the best candidate for NA production. Here, we generated a high-quality chromosome-scale assembly of M. oleifera employing PacBio long-read, short-read Illumina as well as Hi-C sequencing data. The genome assembly consisted of 1.5 Gb with a contig N50 of ~4.9 Mb and a scaffold N50 of ~112.6 Mb. ~98.2% of the assembly was anchored into 13 pseudo-chromosomes. It contains ~1123 Mb repeat sequences, and 27,638 protein-coding genes, 568 tRNAs, 230 rRNAs and 352 other non-coding RNAs. Additionally, we documented candidate genes involved in NA biosynthesis including 20 KCSs, 4 KCRs, 1 HCD and 1 ECR, and profiled the expression patterns of these genes in developing seeds. The high-quality assembly of the genome provides insights into the genome evolution of the M. oleifera genome and candidate genes involved in NA biosynthesis in the seeds of this important woody tree.
Similar content being viewed by others
Background & Summary
Nervonic acid (C24:1 Δ15, NA) is a very long-chain monounsaturated fatty acid, originally isolated in shark brain tissues about 100 years ago1 (Fig. 1). As essential component comprising the white matter and myelin sheath of nerve fibers by combining with sphingosines to form nervonyl sphingolipids, NA has been found to be clinically indispensable and critical in maintaining the brain and nerve cells development and promoting the repair and regeneration of nerve fibers in damaged brain tissues2. With limited availability of marine creatures, NA derived from vegetable oils are alternative resources for supply of pharmaceutical and nutraceutical applications. Till now, NA has been found in 38 plant species belonging to 31 genera and 13 families, among which Malania oleifera has been evaluated to be the best candidate for NA production as M. oleifera has the highest content of NA reported thus far in any seed fat2 (Fig. 1). The M. oleifera genome has been sequenced and is publicly available3, but this assembly is fragmented and does not reach the chromosomal scale, which is impeding genome evolution analysis and genes’ identification and characterization. In the present study, the high-quality chromosome-scale assembly of the M. oleifera genome is provided. Then we further documented key genes involved in the NA biosynthesis by integrating the high quality genome information and transcriptome data.

M. oleifera fruits with a representation of the chemical structure of nervonic acid.
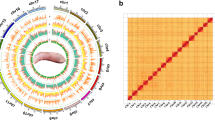
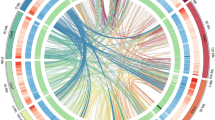
Based on an integrated approach, including PacBio long-read sequencing (51.1 Gb) and short-read Illumina sequencing (135.0 Gb) as well as Hi-C sequencing (173.0 Gb), a chromosome-scale assembly for the M. oleifera genome has been generated. The final M. oleifera genome assembly consisted of 1.5 Gb with a contig N50 of ~4.9 Mb and a scaffold N50 of ~112.6 Mb (Table 1). 98.2% (1468.2 Mb) of the assembly was anchored into 13 pseudo-chromosomes (Figs. 2, 3). The continuity of this genome assembly is significantly higher than that of the previous version, with contig N50 being 4 times higher and scaffold N50 being 24 times higher, and with a significant reduction in gap numbers (596 v.s. 1710; Table 1). Of the assembled genome sequence, 75.1% (1123.1 Mb) were transposable elements with a dominance of long terminal repeats (LTRs), which accounted for 68.0%. The most abundant repeat element families were Copia (28.9%) and Gypsy (30.0%) (Fig. 2, Table 2). We also annotated 27,638 protein-coding genes (33,130 transcripts), 568 tRNAs, 230 rRNAs and 352 other non-coding RNAs (Tables 1, 3). The percentage of complete BUSCOs of genes was 92.2%, with 4.0% missing BUSCOs. The high-quality assembly provides opportunities for documenting key genes involved in the biosynthesis of very long-chain fatty acids including NA (Fig. 4). In total, we documented 20 genes encoding 3-ketoacyl-CoA synthase (KCS), four encoding 3-ketoacyl-CoA reductase (KCR), one encoding 3-hydroxacyl-CoA dehydratase (HCD) and one encoding trans-2, 3-enoyl-CoA reductase (ECR) (Fig. 4). Based on our previous transcriptome data generated from the two stages of M. oleifera seed development4, we revealed that six KCSs, all KCRs, HCD and ECR were expressed in the seeds with a FPKM value > 2 (Fig. 4), suggesting that these key genes were most likely responsible for NA biosynthesis. In conclusion, this high-quality assembly of the M. oleifera genome provides new insights into the evolution of the M. oleifera genome and valuable resources for metabolic engineering for the production of NA in other crops lacking NA.

Circular map of M. oleifera. (1) Density of class I transposable elements, (2) Density of class II transposable elements, (3) Density of protein-coding genes, (4) Density of single nucleotide polymorphisms, (5) GC content, (6) Paralog synteny relationships.

The Hi-C heatmap of chromosome interactions in M. oleifera chromosomes.

The biosynthetic pathway of very long-chain fatty acids including nervonic acid (C24:1, NA) and the expression of key genes in developing seeds (seed1: the initial stage of seed development with 0.88% NA, and seed2: the fast oil accumulation stage with 29.4% NA).
Methods
Sample collection and sequencing
Materials for high-throughput chromosome conformation capture sequencing were taken from a young tree of M. oleifera. The Hi-C library was prepared by Beijing Ori-Gene Science and Technology Co., Ltd (Beijing, China). A total of 700 ng of high molecular weight genomic DNA was cross-linked in situ, extracted, and subsequently digested with a restriction enzyme. The sticky ends of the digested fragments were biotinylated, enriched and sheared to a fragment size of 300–500 bp for preparing the sequencing library. Sequences were obtained from HiSeq X Ten platform (Illumina).
Chromosome-level genome assembly
We started the genome assembly from the previous scaffold-level assembly of M. oleifera3. Gaps of the genome assembly were filled with third generation data using LR_Gapcloser v. 35, run for two rounds to enhance contig continuity. Then pilon v. 1.24 program6 was used to polish one round with second generation data to enhance base-level accuracy. Subsequently the scaffolds were broken into contigs for downstream assembly. For Hi-C data-based assembly, Hi-C data were first mapped to the preprocessed genome using Juicer v. 0.6.87, followed by preliminary Hi-C-assisted chromosome assembly using 3d-DNA v. 1809228, and then the Juicebox module9 for manual adjustment of chromosome boundaries. Then all chromosomes were re-scaffolded one by one with 3d-DNA, and then manually adjusted to correct errors with Juicebox module, including adjusting boundaries, removing wrong insertions and adjusting orientation. LR_Gapcloser v.3 and NextPolish v. 1.3.110 were used for the final optimization. Redundans v. 0.14a11 was used to remove redundancy in un-anchored sequences (identity >= 0.98). We evaluated the step of redundancy removal by mapping Pabio reads (minimap2 v. 2.24 -x map-pb) to the assembly prior to redundancy removal.
The quality and completeness evaluation of the assembled genome
We performed the following four assessments of the new genome: (i) completeness assessment; (ii) redundancy assessment; (iii) assessment of base-level accuracy and heterozygosity rate; and (iv) evaluation of Hi-C interaction. LTR Assembly Index (LAI)12 was examined using LTR_retriever v. 2.9.013. We also assessed the genome using BUSCO v. 5.3.214 with the embryophyta_odb10 lineage dataset for the proportion of intact core genes (both single-copy and multi-copy) and the proportion of missing genes. In the redundancy and heterozygosity assessment, KAT v. 2.4.215 was used by comparing the genome and the corrected Pacbio reads. For the assessment of base-level accuracy and completeness, Merqury v. 1.3 was applied16.
The annotation of the genome
RepeatModeler v. 2.0.117 was employed to generate a repeat sequence library. The intact LTR-RTs were identified using LTR_retriever v. 2.9.013 and clustered to generate another library. Two libraries were merged and repeat regions on the genome were identified using RepeatMasker v. 2.118.
EST and homologous protein evidence for gene annotation were directly used for the 57,298 transcript sequences and 135,881 protein sequences obtained from the genome sketch3. For ab initio gene prediction, the parametric model of AUGUSTUS v. 3.4.019 was trained with single-copy core genes identified by BUSCO v. 3.0.214 and optimized for five rounds. MAKER2 v. 2.31.920 annotation process was first run for one round; then the high quality gene set with Annotation Edit Distance (AED) score less than 0.1 was selected from the annotation results and trained again for AUGUSTUS v. 3.4.019 and perform 5 rounds of optimization, followed by the final round of annotation by MAKER2 v. 2.31.920 without AED selection. Through manual check, we found that there were many false fission genes from MAKER2. Thus, we corrected these issues with gene annotations of the PASA v. 2.4.121 pipeline based on transcript evidence. The completeness of gene annotation was evaluated with BUSCO v. 5.3.214. In addition, tRNA, rRNA and other non-coding RNAs were identified by using tRNAScan-SE22, barrnap (https://github.com/tseemann/barrnap) and RfamScan23, respectively.
The functions of protein-coding genes were annotated based on three strategies: (i) eggNOG-mapper v. 2.0.124 annotation, which annotates the functions of genes by comparing with the eggNOG v. 5.0 homologous gene database, including Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) database. (ii) To identify the best comparison of protein domains, sequence similarity search in which protein sequences were compared with protein databases including Swiss_Prot (https://www.sib.swiss/swiss-prot), TrEMBL (https://www.uniprot.org/statistics/TrEMBL), NR (https://www.ncbi.nlm.nih.gov/refseq/about/nonredundantproteins) and Arabidopsis protein database (https://www.arabidopsis.org/portals/proteome/proteinLocation.jsp) was conducted using diamond v. 2.0.425. The comparison criteria were set >30% and E-value < 1e-5. (iii) Structural domain similarity search: the sub-databases PRINTS, Pfam, SMART, PANTHER, CDD in the InterPro database (www.ebi.ac.uk/interpro) were compared using InterProScan v. 82.026 to obtain the protein information.
Identification and expression patterns of genes involved in the biosynthesis of very long chain fatty acids (VLCFA) from M. oleifera
KCS, KCR, HCD and ECR proteins sequences in Arabidopsis were used as query to search against the protein database of M. oleifera using BLASTP program with e-value > 10−5. All candidate proteins were further confirmed via SMART/Pfam analysis. To profile their expression in seeds, the transcriptome data generated from the two stages of M. oleifera seed development were retrieved from NCBI Sequence Read Archive (SRA) under SRP1584844, and mapping onto the M. oleifera genome using HISAT227 software. Next, the expression level of each gene was calculated and normalized to fragments per kilobase of transcript per million fragments mapped (FPKM) using STRINGTIE28.
Data Records
The Hi-C sequencing data that were used for the genome assembly have been deposited in the NCBI Sequence Read Archive with accession number SRR1830799529 and under BioProject number PRJNA472200. The chromosomal assembly and dataset of gene annotation has been deposited at the Genome Warehouse in National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences/China National Center for Bioinformation under accession number GWHBHOG00000000 (https://ngdc.cncb.ac.cn/gwh/Assembly/24427/show) under BioProject accession number PRJCA008620. The genome assembly has also been deposited at DDBJ/ENA/GenBank under the accession JARUNQ00000000030. The genome annotations have also been deposited at FigShare31.
Technical Validation
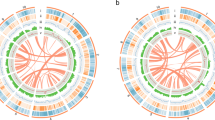
The Hi-C heatmap revealed a well-organized interaction contact pattern along the diagonals within/around the chromosome region (Fig. 3), which indirectly confirmed the accuracy of the chromosome assembly. We evaluated the step of redundancy removal and found that redundancy sequences show a coverage depth peak at the half of that of non-redundancy sequences (Fig. 5), indicating they should be true redundancies from duplicated haplotigs. The k-mer comparison plot revealed high consistency between the final genome assembly and sequencing reads (Fig. 6). The heterozygous rate estimated by KAT was 0.12%. The base-level accuracy (QV) and completeness was 34.9 and 95.9%, respectively, as estimated by Merqury. The LAI index was 8.3. The BUSCO completeness of assembly and annotation was 97.5% and 92.2%, respectively.

Evaluation of the step of redundancy removal.

The k-mer comparison between final genome assembly and sequencing reads.
Code availability
All software and pipelines were executed according to the manual and protocols of the published bioinformatics tools. The version and code/parameters of software have been detailed described in Methods.
References
Tsujimoto, M. & Kimura, K. New fatty acids in shark - liver oil. J. Soc. Chem. 46, 385–388 (1926).
Liu, F. et al. A review of nervonic acid production in plants: prospects for the genetic engineering of high nervonic acid cultivars plants. Front. Plant Sci. 12, 626625 (2021).
Xu, C. et al. Genome sequence of Malania oleifera, a tree with great value for nervonic acid production. GigaScience 8, 1–14 (2019).
Yang, T. et al. Transcriptome analysis reveals crucial genes involved in the biosynthesis of nervonic acid in woody Malania oleifera oilseeds. BMC Plant Biol. 18, 247 (2018).
Xu, G. et al. LR_Gapcloser: a tiling path-based gap closer that uses long reads to complete genome assembly. GigaScience 8, y157 (2018).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. Plos One 9, e112963 (2014).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Hu, J. et al. NextPolish: a fast and efficient genome polishing tool for long read assembly. Bioinformatics 36, 2253–2255 (2020).
Pryszcz, L. P. & Toni, G. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 44, e113 (2016).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126 (2018).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Simão, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Mapleson, D. et al. KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 33, 574–576 (2016).
Rhie, A. et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 1–27 (2020).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. P. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 5, 1–14 (2004).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, 435–439 (2006).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2018).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Kalvari, I. et al. Non‐coding RNA analysis using the Rfam database. Current Protocols in Bioinformatics 62, e51 (2018).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Pertea, M. et al. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR18307995 (2022).
Zhang, R. G. Malania oleifera isolate guangnan, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JARUNQ000000000 (2023).
Zhang, R. G. Genome annotations of Malania oleifera. Figshare https://doi.org/10.6084/m9.figshare.22580476 (2023).
Acknowledgements
We thank Dr. Jane Marczewski for revision of the paper. This work was supported by the Key Project of Natural Science Foundation of Yunnan Province (Grant No. 202001AS070019), CAS “Light of West China” Program, Reserve Talents for Academic and Technical Leaders of Middle-aged and Young People in Yunnan Province (Grant No. 2018HB066) and Ten Thousand Talent Program of Yunnan Province (Grant No. YNWR-QNBJ-2018-174).
Author information
Authors and Affiliations
Contributions
Y.M. W.X. and W.S. designed the study. T.Y., R.Z., X.T., G.Y., Y.S. and S.W. performed the experiments and analysed the data. Y.M. W.X. and W.S. wrote the paper. S.W., J.M. and A.L. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, T., Zhang, R., Tian, X. et al. The chromosome-level genome assembly and genes involved in biosynthesis of nervonic acid of Malania oleifera. Sci Data 10, 298 (2023). https://doi.org/10.1038/s41597-023-02218-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-023-02218-8
