
Abstract
Understanding of adaptation and evolution of organisms in the deep sea requires more genomic resources. Zoroaster cf. ophiactis is a sea star in the family Zoroasteridae occurring exclusively in the deep sea. In this study, a chromosome-level genome assembly for Z. cf. ophiactis was generated by combining Nanopore long-read, Illumina short-read, and Hi-C sequencing data. The final assembly was 1,002.0 Mb in length, with a contig N50 of 376 Kb and a scaffold N50 of 40.4 Mb, and included 22 pseudo-chromosomes, covering 92.3% of the assembly. Completeness analysis evaluated with BUSCO revealed that 95.91% of the metazoan conserved genes were complete. Additionally, 39,426 protein-coding genes were annotated for this assembly. This chromosome-level genome assembly represents the first high-quality genome for the deep-sea Asteroidea, and will provide a valuable resource for future studies on evolution and adaptation of deep-sea echinoderms.
Similar content being viewed by others


Background & Summary
Sea stars or starfish, members of the class Asteroidea, are one of the five extant groups within Echinodermata. Asteroids are a diverse group including about 1,900 extant species1. Asteroids occur worldwide in various marine habitats from the intertidal to the hadal zone (~10,000 m)2. As major predators, asteroids play important roles in marine ecosystems by affecting the ecology of the prey and the community structure3. With a long fossil record, sea stars are of tremendous interest of paleontologists and evolutionary biologists4. The remarkable life history diversity in the Asteroidea make them good subjects for studies of evolutionary developmental biology, developmental ecology and regeneration5,6,7.
Of all the extant starfish families, approximately half occur exclusively in the deep sea (>200 m), and many families among others also comprise deep-sea members1, suggesting a high diverse of asteroids in the deep-sea floor. Sea stars of the family Zoroasteridae (order Forcipulatida), occurring exclusively in the deep sea (~200–6,000 m), are prominent members of the deep-sea benthic animals, and they are often collected in high densities, suggesting their potentially important roles in the deep-sea ecosystems8. Zoroasteridae includes seven genera and approximately 40 species, and is phylogenetically basal among Forcipulatida1, implying an important evolutionary role of this asteroid group.
It is well known that the deep sea is a unique environment that is mostly characterized by darkness, low temperatures, high hydrostatic pressure and limited food resources9. The harsh environment in the deep sea challenges organisms living there. Recently, several deep-sea animal species, such as sea cucumber10, marine mussel11, limpet12, cold-water coral13, anemone14, tubeworms15,16,17 and fish18,19, have been investigated through the genomic data, demonstrating molecular mechanisms of adaptation to the deep sea. As one of the main members of the sea floor, however, genomic resources for the diverse starfish at the chromosome level are scarce20,21,22,23,24,25, and no genomic resources has been available up to now for the deep-sea starfish, which hinders studies on their evolution, speciation and adaptation to the deep sea.
In the present study, we present a chromosome-level genome assembly for the deep-sea starfish, Zoroaster cf. ophiactis, the first high-quality genome assembly for the deep-sea Asteroidea. The species, belonging to the deep-sea asteroid family Zoroasteridae, was collected at depth of 1,750 m in the South China Sea. A combined strategy involving Nanopore long-read, Illumina short-read and Hi-C sequencing technologies was used in this study. This high-quality genome will serve as a valuable resource for future studies on the adaption and evolution of deep-sea starfish.
Methods
Sample collection
One specimen of the starfish Z. cf. ophiactis was collected in the northern South China Sea (111.033E, 17.597 N, 1750 m in depth) by the manned submersible Shenhai Yonghshi during the cruise TS07 of R/V Tansuo 1 in 2018. Tissues of one arm were frozen with liquid nitrogen and then kept at −80 °C until further use.
DNA extraction, library preparation and sequencing
High molecular weight (HMW) genomic DNA was extracted from the frozen tissues by using the SDS method and then purified with the QIAGEN® Genomic kit (QIAGEN) following the manufacturer’s instructions. The quality of the extracted DNA was assessed using 1% agarose gel electrophoresis, and NanoDrop™ One UV-Vis spectrophotometer (Thermo Fisher Scientific, USA) with the OD 260/280 of 1.8–2.0 and OD 260/230 of 2.0–2.2. The quantity of the DNA was measured by Qubit® 3.0 Fluorometer (Invitrogen, USA). DNA libraries for Illumina sequencing were prepared using the Truseq Nano DNA HT Sample Preparation Kit (Illumina USA) according to the manufacturer’s protocols. The libraries were sequenced on the Illumina Hiseq 4000 platform, yielding 150-bp paired-end reads with an insert size of ~350 bp. In total, ~103 Gb of Illumina raw reads were obtained. For the Oxford Nanopore library preparation, genomic DNA fragments > 20 kb were selected using the BluePippin system (Sage Science, USA). Approximate 2 µg HMW DNA was used as input material, according to the manufacturer’s instructions, for the ligation Sequencing kit SQK-LSK109 (Oxford Nanopore Technologies, UK). Sequencing was performed on a Nanopore PromethION sequencer (Oxford Nanopore Technologies, UK). A total of ~60 Gb of Nanopore raw reads were generated. A high-throughput chromatin conformation capture (Hi-C) method was applied to generate a chromosome-level genome. Briefly, the frozen arm tissues were crosslinked with 2% formaldehyde, and then digested with the restriction enzyme MboI (400 units). The DNA ends were tagged with the biotin-14-dCTP and fragments were sheared to 200–600 bp. The resulting Hi-C library was sequenced on the Illumina HiSeq 4000 platform (paired-end 150 bp reads). A final ~72 Gb of raw reads were obtained.
RNA extraction and transcriptome sequencing
The total RNA was isolated from the frozen arm tissue using Trizol (Invitrogen, Carlsbad, CA, USA), following the manufacturer’s instructions. Concentration of the isolated RNA was measured using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA), and its quality was evaluated by 1.5% agarose gel electrophoresis. RNA integrity was quantified by the Agilent 5400 fragment analyzer (Agilent, USA). RNA-seq libraries were constructed by the NEBNext® Ultra™ RNA Library Prep Kit (NEB, USA) following the manufacturer’s instructions. Libraries were then sequenced on an Illumina Hiseq 4000 platform (paired-end 150 bp reads). A total of ~8 Gb raw reads were yielded and used for the gene prediction.
Genome assembly
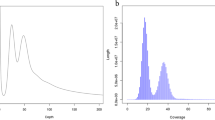
Genome size, proportion of repetitive sequences and heterozygosity was estimated by using the Illumina short-read data and the k-mer analysis with Jellyfish v2.3.026. Based on the ~103 Gb Illumina data and the 19-mer frequency distribution analysis, a total of 78,106,733,386 k-mers were obtained after removing k-mers with abnormal depth, and the 19-mer peak was at a depth of 74. Therefore, the genome size of Z. cf. ophiactis was estimated to be 78,106,733,386/74 = 1,055 Mb, the heterozygosity was about 0.32%, and the proportion of repetitive sequences was roughly 69.85% (Fig. 1).

K-mer distribution (K = 19) of Zoroaster cf. ophiacti genome. The x-axis is the k-mer depth, and the y-axis represents the corresponding frequency of the k-mer at a given depth.
The Nanopore long-read data were used to generate a contig-level assembly for the Z. cf. ophiactis genome. A preliminary assembly was generated by using the program WTDBG2 v2.527 (parameters: -p 19 -AS 2 -s 0.05 -L 5000 -t 36 -fo starfish). Then, three rounds of polishing were carried out with ~103 Gb of Illumina reads by the software Nextpolish v1.2.028. The Hi-C technology was used for chromosome-level genome assembly. Raw Hi-C paired reads were trimmed by Fastp v0.20.029, and aligned to the draft assembly with Juicer v1.5.730 using default settings. Contigs were scaffolded using 3D-DNA pipeline v18011431 with all valid Hi-C reads. The chromosome-scale scaffolds were adjusted manually using Juicebox v1.11.081232 with the aid of the Hi-C contact map whereby redundant contigs and misjoins were removed and fixed. All the corrections were incorporated into the assembly using the 3D-DNA post-review pipeline. Ultimately, the contigs were anchored to 22 pseudo-chromosomes, accounting to 92.3% of the total genome (Table 1; Fig. 2). The lengths of the 22 pseudo-chromosomes ranged from 22.2 Mb to 107.2 Mb (Table 2). The final assembly was 1,002.0 Mb in length, containing 8,895 congtigs with N50 of 376 kb and 616 scaffolds with scaffold N50 of 40.4 Mb (Table 1).

Characteristics of the genome assembly of Zoroaster cf. ophiactis. (a) Genome overview of the 22 chromosomes. Tracks from inner to outer represent repeats coverage (19–96%), genes density (1–85), GC content (35–47%), genome sequence depth (7–100 X), and assembled chromosomes, respectively, with densities calculated within a 500-kb window. (b) Hi-C contact map produced by 3D-DNA. The blue square represents a pseudo-chromosome, and small green squares inside each blue square are contigs that make up the chromosome.
Repeat annotation
Repetitive elements in the genome assembly were annotated by using RepeatModeler v2.0.133, RepeatMasker v4.0.734 and TRF v4.0.935. Ultimately, a total of 673.3 Mb repeat sequences were identified, accounting for 63.9% of the whole genome. The DNA elements (114.3 Mb) were the predominant type of the transposable elements (TEs), which represented 10.84% of the genome, followed by the long interspersed nuclear elements (LINEs) with the portion of 7.69% in the genome. The short interspersed nuclear elements (SINEs) and the long terminal repeat (LTR) retrotransposons occupied 2.86% and 2.95% of the genome, respectively.
Gene prediction and annotation
Protein-coding genes were predicted with three different strategies: ab initio gene prediction, homology-based prediction, and transcript prediction. The ab initio gene prediction was performed using Augustus v2.436, and GlimmerHMM v3.0.437. For the homology-based prediction, protein sequences from five echinoderm species, Acanthaster planci (GCF_001949145.1)38, Anneissia japonica (GCF_011630105.1)39, Apostichopus japonicus (GCA_002754855.1)40, Lytechinus variegatus (GCF_018143015.1)41 and Strongylocentrotus purpuratus (GCF_000002235.5)42, were downloaded from the NCBI database for the gene prediction as implemented in TblastN v2.2.2943 with an e-value ≤ 1e-5. For the transcriptome-based annotation, clean RNA-seq reads were aligned to the Z. cf. ophiactis genome assembly by using HISAT2 v2.2.144, and gene set was predicted by using PASA v2.3.245 pipeline. Finally, results from ab initio prediction, homology-based prediction, and transcript prediction were integrated by using EvidenceModeler v1.1.146 to generate a consensus and non-redundant gene set. Overall, 39,426 protein-coding genes were annotated for the Z. cf. ophiactis genome by combining three different methods, with an average of exon and intron length of 217.7 bp and 1952.8 bp, respectively (Table 3). The average length and number of the genes, exons, and introns of the Z. cf. ophiactis genome were comparable to those reported in other sea stars24.
Functional annotation for the predicted protein-coding genes was performed against six public databases, Kyoto Encyclopedia of Genes and Genomes (KEGG), Gene Ontology (GO), NCBI-NR (non-redundant protein database), Swiss-Prot, SMART and InterProScan with BLASTP v2.2.2347 and an e-value cutoff of 1e-5. The results showed that 36,557 (92.72%) predicted genes were annotated by at least one public database (Table 4).
Data Records
All the raw sequencing data of Illumina, Nanopore, and Hi-C obtained in this study have been deposited in the NCBI Sequence Read Archive (SRA) database with the accession numbers SRR22953576- SRR22953579, and SRR24759671 under the BioProject PRJNA89147948. The final genome assembly has been deposited in the Science Data Bank of Chinese Academy of Sciences49 and the GenBank database under the accession number JAQQFT01000000050. Files of genome annotation, repeat annotation, gene functional annotation and gene family expansion have been submitted to the Figshare database51.
Technical Validation
Assessment of genome assembly
The genome size of Z. cf. ophiactis was estimated to be about 1,055 Mb based on the 19-mer frequency distribution analysis. The estimation of genome length was consistent with our final genome assembly (1,002 Mb, Table 1). It is noted that the Z. cf. ophiactis genome assembly is much larger than genomes reported for other asteroids, including species in the order Forcipulatida (402–561 Mb)21,22,23,24, and those in the other order, Valvatida (384–608 Mb)20,25,52. In addition, 22 pseudo-chromosomes were generated for the Z. cf. ophiactis genome assembly. The chromosome number is consistent with previous karyotyping studies on some asteroids, including species from Forcipulatida53. This is also proved by recent genome studies on several starfish species where 22 pseudo-chromosomes were identified by the Hi-C method22,23,24.
To assess the accuracy of Z. cf. ophiactis genome assembly, the completeness of the genome assembly was assessed using the conserved metazoan gene set “metazoan_odb10” from the Benchmarking Universal Single-Copy Orthologs (BUSCO) v4.054. The genome assembly was found to have a high level of completeness (95.91%). Of the 954 single-copy orthologs, 95.28% were complete and single-copy, 0.63% complete and duplicated, 0.84% fragmented, and 3.25% were missing (Table 5). In addition, clean Illumina short reads used for the genome survey were aligned back to the Z. cf. ophiactis genome assembly with Burrows-Wheeler aligner (BWA) v0.7.17-r119855. As a result, 99.35% of the short reads were mapped to the genome. Together, these results indicate the high quality of the Z. cf. ophiactis genome assembly.
Chromosome synteny
Syntenic relationships among the genomes of Z. cf. ophiactis and the other two Forcipulatida star fish, Asterias rubens (GCF_902459465.1)56 and Plazaster borealis (GCA_021014325.1)24 were inferred and visualization by Blastp and NGenomeSyn v1.3757. The three starfish appeared to have very conserved syntenic relationships as every chromosome matched each other well (Fig. 3). This finding provides new evidence of a high level of synteny conservation in the order Forcipulatida24.

Chromosomal synteny among genomes of Zoroaster cf. ophiactis and the other two starfish (Asterias rubens, and Plazaster borealis) in the order Forcipulatida. Numbers in the rectangles represent chromosomes of each genome.
Gene annotation validation
To evaluate the completeness of the annotated gene set, we performed the BUSCO analysis using the conserved metazoan database “metazoan_odb10”. The results revealed that 97.07% of the conserved single copy ortholog genes to be complete (96.23% single-copied genes and 0.84% duplicated genes), 0.73% fragmented and 2.2% missing (Table 5). Additionally, functional annotation of the predicted genes revealed that 92.72% of them were annotated by at least one public database (Table 4).
Phylogenetic relationships among Z. cf. ophiactis and other eight echinoderm species, including Asterias rubens (GCF_902459465.1)56, Plazaster borealis (GCA_021014325.1)24, Acanthaster planci (GCF_001949145.1)38, Patiria miniata (GCF_015706575.1)58, Apostichopus japonicus (GCA_002754855.1)40, Strongylocentrotus purpuratus (GCF_000002235.5)42, Lytechinus variegatus (GCF_018143015.1)41, Anneissia japonica (GCF_011630105.1)39, were inferred by using the maximum likelihood (ML) method. Homo sapiens (GCF_000001405.39)59 was used as the outgroup. Single-copy orthologs among genomes of all species were determined using OrthoFinder v2.3.360 with the default parameters. Multiple alignments of the protein sequences were performed with Muscle v3.8.155161. RAxML v8.2.1262 was used to produce the ML trees with the following parameters: -m GTRGAMMA -x 12345 -N 100. The phylogenetic tree was reconstructed with 1,316 single-copy orthologs (Fig. 4). Zoroaster cf. ophiactis was clustered with A. rubens and P. borealis within the family Asteriidae, where they all belong to the order Forcipulatida, and then were grouped with two starfish species (A. planci and P. miniata) from the order Valvatida. Expansion and contraction of gene families were evaluated by CAFE v563 with a p-value of 0.05. A total of 1,162 gene families were expanded while 55 were contracted in the deep-sea starfish, Z. cf. ophiactis (Fig. 4).

Maximum likelihood phylogenetic tree of Zoroaster cf. ophiactis and other eight echinoderms. Bootstrap support values for all the nodes are equal to 100. Numbers of expanded (red) and contracted (blue) gene families are shown.
Code availability
No specific code was used in this study. All commands and pipelines used in the data processing were performed according to manuals and protocols of corresponding bioinformatics software, with parameters described in the Methods section. If no detailed parameters were mentioned for a software, default parameters were used.
References
Mah, C. L. & Blake, D. B. Global diversity and phylogeny of the Asteroidea (Echinodermata). PLoS One 7, e35644 (2012).
Jamieson, A. The Hadal Zone: Life In The Deepest Oceans (Cambridge University Press, 2015).
Gaymer, C. F. & Himmelman, J. H. A keystone predatory sea star in the intertidal zone is controlled by a higher-order predatory sea star in the subtidal zone. Mar. Ecol. Prog. Ser. 370, 143–153 (2008).
Lawrence, J. M. Starfish: Biology And Ecology Of The Asteroidea (Johns Hopkins University Press, 2013).
Byrne, M. in Starfish: Biology And Ecology Of The Asteroidea (ed. Lawrence, J. M.) Ch. 5 (Johns Hopkins University Press, 2013).
Byrne, M. et al. Transcriptomic analysis of sea star development through metamorphosis to the highly derived pentameral body plan with a focus on neural transcription factors. DNA Res. 27, dsaa007 (2020).
Meyer, A. & Hinman, V. in Current Topics in Developmental Biology Vol. 147 (ed. Goldstein, B. & Srivastava, M.) Ch. 18 (Academic Press, 2022).
Mah, C. Phylogeny of the Zoroasteridae (Zorocallina; Forcipulatida): evolutionary events in deep-sea Asteroidea displaying Palaeozoic features. Zool. J. Linn. Soc. 150, 177–210 (2007).
Danovaro, R., Snelgrove, P. V. & Tyler, P. Challenging the paradigms of deep-sea ecology. Trends Ecol. Evol. 29, 465–475 (2014).
Liu, R., Liu, J. & Zhang, H. Positive selection analysis provides insights into the deep-sea adaptation of a hadal sea cucumber (Paelopatides sp.) to the Mariana Trench. J. Oceanol. Limnol. 39, 266–281 (2021).
Sun, J. et al. Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat. Ecol. Evol. 1, 0121 (2017).
Liu, R. et al. De novo genome assembly of limpet Bathyacmaea lactea (Gastropoda: Pectinodontidae), the first reference genome of a deep-sea gastropod endemic to cold seeps. Genome Biol. Evol. 12, 905–910 (2020).
Zhou, Y. et al. The first draft genome of a cold-water coral Trachythela sp. (Alcyonacea: Stolonifera: Clavulariidae). Genome Biol. Evol. 13, evaa265 (2021).
Feng, C. et al. The genome of a new anemone species (Actiniaria: Hormathiidae) provides insights into deep-sea adaptation. Deep Sea Res. Part I Oceanogr. Res. Pap. 170, 103492 (2021).
Li, Y. et al. Genomic adaptations to chemosymbiosis in the deep-sea seep-dwelling tubeworm Lamellibrachia luymesi. BMC Biol. 17, 1–14 (2019).
Sun, Y. et al. Genomic signatures supporting the symbiosis and formation of chitinous tube in the deep-sea tubeworm Paraescarpia echinospica. Mol. Biol. Evol. 38, 4116–4134 (2021).
de Oliveira, A. L., Mitchell, J., Girguis, P. & Bright, M. Novel insights on obligate symbiont lifestyle and adaptation to chemosynthetic environment as revealed by the giant tubeworm genome. Mol. Biol. Evol. 39, msab347 (2022).
Wang, K. et al. Morphology and genome of a snailfish from the Mariana Trench provide insights into deep-sea adaptation. Nat. Ecol. Evol. 3, 823–833 (2019).
Mu, Y. et al. Whole genome sequencing of a snailfish from the Yap Trench (~7,000 m) clarifies the molecular mechanisms underlying adaptation to the deep sea. PLoS Genet. 17, e1009530 (2021).
Hall, M. R. et al. The crown-of-thorns starfish genome as a guide for biocontrol of this coral reef pest. Nature 544, 231–234 (2017).
Ruiz‐Ramos, D. V., Schiebelhut, L. M., Hoff, K. J., Wares, J. P. & Dawson, M. N. An initial comparative genomic autopsy of wasting disease in sea stars. Mol. Ecol. 29, 1087–1102 (2020).
Lawniczak, M. K. & Consortium, D. T. O. L. The genome sequence of the spiny starfish, Marthasterias glacialis (Linnaeus, 1758). Wellcome Open Res. 6, 295 (2021).
DeBiasse, M. B. et al. A chromosome-level reference genome for the giant pink sea star, Pisaster brevispinus, a species severely impacted by wasting. J. Hered. 113, 689–698 (2022).
Lee, Y. et al. Chromosome-level genome assembly of Plazaster borealis sheds light on the morphogenesis of multiarmed starfish and its regenerative capacity. GigaScience 11, giac063 (2022).
Yuasa, H. et al. Elucidation of the speciation history of three sister species of crown-of-thorns starfish (Acanthaster spp.) based on genomic analysis. DNA Res. 28, dsab012 (2021).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158 (2020).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25, 4.10.11–14.10.14 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Stanke, M., Schöffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 1–11 (2006).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Baughman, K. W. et al. Acanthaster planci, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:BDGF01000000 (2016).
Li, Y. et al. Genomic insights of body plan transitions from bilateral to pentameral symmetry in echinoderms. Communications Biology 3, 371 (2020).
Zhang, X. et al. The sea cucumber genome provides insights into morphological evolution and visceral regeneration. PLoS Biol. 15, e2003790 (2017).
Davidson, P. L. et al. Chromosomal-level genome assembly of the sea urchin Lytechinus variegatus substantially improves functional genomic analyses. Genome Biol. Evol. 12, 1080–1086 (2020).
Sodergren, E. et al. The genome of the sea urchin Strongylocentrotus purpuratus. Science 314, 941–952 (2006).
Altschul, S., Gish, W., Miller, W., Myers, E. & Lipman, D. Basic local aligment search tool. J. Mol. Biol. 215, 403–410 (1990).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, 1–22 (2008).
Kent, W. J. BLAT—the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
NCBI BioProject https://identifiers.org/ncbi/insdc.sra:SRP415507 (2023).
Zhou, Y., Liu, J. & Zhang, H. Chromosome-level genome of a deep-sea starfish, Zoroaster sp., provides insights into the adaption of Asteroidea to the deep sea. Science Data Bank https://doi.org/10.57760/sciencedb.04022 (2022).
Liu, J., Zhou, Y., Pu, Y. & Zhang, H. Zoroaster sp. YZ-2022 isolate SQW42HX01, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAQQFT010000000 (2023).
Liu, J. & Zhou, Y. Chromosome-level genome assembly of Zoroaster sp. Figshare https://doi.org/10.6084/m9.figshare.21780527.v1 (2023).
Arshinoff, B. I. et al. Echinobase: leveraging an extant model organism database to build a knowledgebase supporting research on the genomics and biology of echinoderms. Nucleic Acids Res. 50, D970–D979 (2022).
Saotome, K. & Komatsu, M. Chromosomes of Japanese starfishes. Zool. Sci. 19, 1095–1103 (2002).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Wellcome Sanger Institute. Asterias rubens, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:CABPRM030000000 (2019).
He, W. et al. NGenomeSyn: an easy-to-use and flexible tool for publication-ready visualization of syntenic relationships across multiple genomes. Bioinformatics 39, btad121 (2023).
Ku, C. J., Cary, G. A. & Hinman, V. F. Patiria miniata isolate m_02_andy, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JADOBP010000000 (2020).
NCBI Assembly https://identifiers.org/insdc.gca:GCF_000001405.39 (2019).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157 (2015).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Mendes, F. K., Vanderpool, D., Fulton, B. & Hahn, M. W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518 (2020).
Acknowledgements
We are grateful to the captain, crew and scientific staffs on the R/V Tansuo 1, and the manned submersible Shenhai Yongshi. We also thank Dr. Christopher Mah from National Museum of Natural History, Smithsonian Institution (Washington D.C., USA), and Dr. Ruiyan Zhang from Second Institute of Oceanography, Ministry of Natural Resources (Hangzhou, China), for help in species identification. This work was supported by the major scientific and technological projects of Hainan Province (ZDKJ2019011), National Natural Science Foundation of China (42106121), Strategic Priority Research Program of the Chinese Academy of Sciences (CAS) (XDA22050303).
Author information
Authors and Affiliations
Contributions
H.Z. and J.L. conceived the study. H.Z. collected the sample. Y.Z. conducted bioinformatic analyses. J.L. wrote the manuscript. J.L. and Y.Z. interpreted the data and prepared the figures. Y.P. collected public genomic data. All the authors revised and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, J., Zhou, Y., Pu, Y. et al. A chromosome-level genome assembly of a deep-sea starfish (Zoroaster cf. ophiactis). Sci Data 10, 506 (2023). https://doi.org/10.1038/s41597-023-02397-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-023-02397-4
This article is cited by
-
Chromosome-level genome assembly of the northern Pacific seastar Asterias amurensis
Scientific Data (2023)
