
Abstract
Transferable and mechanistic understanding of cross-scale interactions is necessary to predict how coastal systems respond to global change. Cohesive datasets across geographically distributed sites can be used to examine how transferable a mechanistic understanding of coastal ecosystem control points is. To address the above research objectives, data were collected by the EXploration of Coastal Hydrobiogeochemistry Across a Network of Gradients and Experiments (EXCHANGE) Consortium – a regionally distributed network of researchers that collaborated on experimental design, methodology, collection, analysis, and publication. The EXCHANGE Consortium collected samples from 52 coastal terrestrial-aquatic interfaces (TAIs) during Fall of 2021. At each TAI, samples collected include soils from across a transverse elevation gradient (i.e., coastal upland forest, transitional forest, and wetland soils), surface waters, and nearshore sediments across research sites in the Great Lakes and Mid-Atlantic regions (Chesapeake and Delaware Bays) of the continental USA. The first campaign measures surface water quality parameters, bulk geochemical parameters on water, soil, and sediment samples, and physicochemical parameters of sediment and soil.
Similar content being viewed by others
Background & Summary
The structure and function of coastal ecosystems vary considerably across relatively small spatial scales, resulting in dynamic hydrological and biogeochemical behaviors along the gradient of coastal upland, wetland, and surface water environments1,2. Insight into drivers of spatial heterogeneity can be elucidated by linking biogeochemical data with ecosystem properties3,4, enabling scientific discovery and model parameterization, such as furthering mechanistic understanding of coastal ecosystems and improving uncertainty constraints of coastal models1,5.
Open access and interoperable coastal biogeochemical datasets are needed to predict how coastal systems will respond to global change3,6. The Great Lakes and Mid-Atlantic regions have a wealth of long-term monitoring programs hosting open access datasets, such as the National Estuarine Research Reserve7, the Great Lakes Wetland Monitoring Program8, and the Chesapeake Bay Program9, among others. However, the synthesis of existing data streams across traditional ecosystem and disciplinary boundaries is still relatively sparse1,5. Here, we describe datasets collected as part of EXCHANGE Campaign 1 (EC1), which establishes a baseline understanding of the chemical forms and distribution of carbon and nutrients across coastal terrestrial-aquatic interface (TAI) research sites in the Great Lakes and Mid-Atlantic regions (Chesapeake and Delaware Bays) of the continental USA that can be utilized to conduct synthesis. EXCHANGE adds to existing efforts in these regions by developing a consortium of regional researchers interested in exchanging knowledge and information, with a molecular level focus that spans upland to aquatic domains, that can contribute to understanding of how coastal systems will respond to global change.
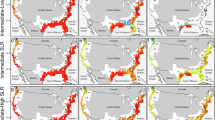
In the Fall of 2021, the EXCHANGE Consortium collected samples from 52 coastal terrestrial-aquatic interfaces (TAIs). At each of these TAI sites, the consortium collected soil samples from across a transverse elevation gradient, which included soils from coastal upland forests, transitional forests, and wetlands. The consortium also collected surface water and nearshore sediment samples adjacent to the transverse elevation gradient (Fig. 1). Samples collected from EC1 were analyzed for bulk geochemical parameters, bulk physicochemical parameters, organic matter characteristics, and redox-sensitive elements. These datasets are useful in evaluating the physicochemical factors that drive spatial variations in the cycling of organic matter across coastal terrestrial-aquatic interfaces (TAIs). They also facilitate an understanding of the biogeochemical control points in coastal ecosystems that can be assessed for their transferability across different coastal systems. Here, we describe version one (v1) of the key baseline datasets that are currently published open access10. We also describe additional datasets that will be published in subsequent versioning of the data package in the Supplementary Information.

EXCHANGE Campaign 1 sites were located in the Great Lakes and Mid-Atlantic Regions. 52 terrestrial-aquatic interfaces were sampled, from uplands to nearby waters (lake, estuary, stream, river, etc) for surface soils, sediments, and water samples.
Methods
Sampling and processing
Sampling design
The experimental design of EC1 was developed via workshops (following open science principles11) from conception to data analysis and publication. Coastal researchers gathered virtually to design a spatially distributed sampling campaign across Great Lakes and Mid-Atlantic regions (Fig. 1). The EC1 consortium collected surface waters, soils, sediments, and site level metadata using standardized sampling kits. Following sample collection, all sample kits were shipped to the Marine and Coastal Research Laboratory (Sequim, WA), part of Pacific Northwest National Laboratory.
Site metadata
At each site, the EXCHANGE consortium collected standardized site metadata, such as latitude, longitude, and type of water system (e.g., estuary, lake). Additional site metadata, such as elevation and soil type, were extracted from publicly available databases (e.g., GoogleEarth) using site coordinates.
Surface waters
Field-filtered water – using 0.22 µm Sterivex syringe filters – was collected in vials for dissolved organic carbon (DOC), total dissolved nitrogen (TDN), common dissolved ions, stable water isotopes, and several organic matter characterization methods. Samples were filtered into vials in the field and preserved by freezing or storing at 4 °C until analyzed, depending on the analyte (Table 1). A 125 mL amber HDPE bottle of unfiltered water was collected with no headspace for pH, oxygen-reduction potential (ORP), alkalinity, and conductivity measurements of the surface waters. Unfiltered surface water samples were also collected in 1 L acid-cleaned HDPE amber bottles for total suspended solids and filtered to 0.2 µm in the lab, within 48 hours of collection. Lab-filtered 1 L grab samples were extracted for several organic matter characterization methods (e.g., high-resolution mass spectrometry) using standard solid phase extraction (SPE) procedures12. The filtered samples were stored at 4 °C until SPEs were completed, within 2 weeks of sample collection. Briefly, SPE was performed by passing one liter of sample through a 6 mL/1 g PPL SPE cartridge (Agilent PPL), after being acidified 24 hours before extraction to a pH of 2. Samples were then eluted in LC-MS grade methanol and were stored at −20 °C until analysis. Additional analysis beyond those reported herein (e.g. common dissolved ions and stable water isotopes) will be performed on archived filtered waters or SPE extracts as appropriate for the analysis method, and appended to future versions of the data package10.
Soils
Surface soils (top 5 cm of soil profile) were collected from the three transect locations (upland, transition, and wetland) from each TAI site. Soils were collected as intact cores (using HYPROP sampling rings, 5 cm diameter × 5 cm depth) and as surface grab samples (using 2.5 oz plastic (clear polypropylene) jars and plastic bags). The intact cores were refrigerated at 4 °C upon arrival to the laboratory. Subsamples of soil grabs samples were either immediately processed, frozen (−20 °C), or refrigerated, based on the analyses planned (Table 1). Frozen grab samples were freeze-dried, catalogued, and sieved to 5.6 mm before additional analyses (Supplementary Information; Figure S1; Table S1). Water retention curves, particle size analysis, X-ray absorption spectroscopy measurements, and any other additional analysis that will be performed will be appended to future versions of the data package (methods outlined in Supplementary Information)10.
Sediments
Surface sediments (i.e., top 5–10 cm of sediment) were collected into clear 2.5 oz polypropylene jars and frozen at −20 °C upon arrival for archival purposes. One full plastic bag of sediment was also collected for gravimetric water content (GWC) and was stored at 4 °C until analysis. Immediately upon arrival, subsamples from the sealed plastic bags were collected in minimal oxygen conditions and frozen for Fe X-ray absorption fine structure (XAFS) analysis. X-ray absorption spectroscopy measurements and analyses performed on sediment samples will be appended to future versions of the data package (methods outlined in Supplementary Information)10.
Surface water analyses
Common water quality measurements (pH, ORP, conductivity, alkalinity)
Common water quality measurements (i.e., pH, ORP, conductivity, alkalinity) were performed on unfiltered surface water samples, within 24 hours of receiving. Samples were measured simultaneously for temperature, specific conductivity, oxidation-reduction potential, and alkalinity using a Mettler Toledo T7 auto-titrator equipped with an auto-sampler. Prior to starting each run and after every five samples, conductivity and pH sensors were checked with standards, and were recalibrated if outside the acceptable tolerance (+/− 1% for conductivity, and +/− 0.05 for pH). Conductivity was calibrated with a 50,000 μS/cm (+/− 1%) solution to cover the salinity range represented by samples (0 to ~35 PSU). pH was calibrated using a three-point calibration curve (using calibration solutions of pH 4.01, 7.00, and 10.00). Alkalinity was determined by titration with 0.02 N HCl to an endpoint of pH 4.00, following standard United States Geological Survey (USGS) procedures13. All water quality variables underwent quality control to flag values outside of sensor analytical ranges.
Dissolved organic carbon and total dissolved nitrogen
Field-filtered samples were stored at 4 °C until analyzed for dissolved organic carbon (DOC) and total dissolved nitrogen (TDN). DOC and TDN analyses were performed simultaneously, within one week of sample collection on a Total Organic Carbon Analyzer (Shimadzu TOC-L). DOC was measured as the best 3 of 5 injections, after in-line acidification with 1:12 hydrochloric acid, as non-purgeable organic carbon (NPOC) via catalytic combustion. TDN was measured by chemiluminescence as the best 3 of 5 injections. A combined carbon and nitrogen check standard was run every 10 samples; all values were within 10 ± 7% of the concentration for DOC and 6.5 ± 7% of the concentration for TDN. Peaks were disregarded if the coefficient of variation between replicate injections was greater than 2.0%. Data underwent quality control, including visual inspection of calibration curves, check standards, and sample peak shapes; values were flagged when they were outside of the calibration curve and instrument detection limit ranges.
Total suspended solids
Total suspended solids were measured on 1 L grab samples and filtered within 24 hours of sample collection, following Environmental Protection Agency (EPA) method 160.214 with slight modifications. Samples were filtered through pre-combusted and pre-weighed glass fiber filters (GFF, nominal pore size of 0.7 µm). The filtrate was then filtered through 0.2 µm PES filters and stored at 4 °C until solid phase extraction (SPE) procedures were performed.
GF filters were dried in a 45 °C oven for 24–72 hours for TSS. Filters were dried until the filter mass was stable and stored in a desiccator for 24–48 hours after drying, until final weights were taken. Process blanks were filtered concurrently with sample filtering, and average blank signal was below detection. TSS were calculated gravimetrically as follows:
The volume filtered in mL was determined via mass and corrected for density of variable salinity waters using temperature, pressure and salinity data obtained from the titrator dataset with the package gsw15 in R version 4.2.1. When the common water quality measurements samples were not collected at a site, data were gap filled by taking the average of all adjacent kits. Data underwent further quality control to flag values below the blank and above the reported method detection limit for the EPA method14.
Colored dissolved organic matter absorbance and fluorescence
UV absorbance scans and excitation-emission matrices (EEMs) were collected simultaneously with an Aqualog (Horiba Scientific) on filtered sub-samples, which were stored at 4 °C until analysis. Absorbance was measured from 230 to 800 nm in 3 nm intervals, and blank corrected prior to exporting the data. EEMs were collected with the same wavelength constraints and further processed with drEEM toolbox v. 6.0 for Matlab16 (https://www.openfluor.org). EEMs processing included blank correction, inner filter correction17, and normalization to Raman Scatter units based on daily water Raman scans collected at an excitation of 350 nm.
High-resolution mass spectrometry
Aliquots of SPE extracts described in the sampling and processing methods for surface waters were normalized to a DOC concentration of 50 mg C/L prior to FTICR-MS analysis18. Spectra were collected at the Environmental Molecular Sciences Laboratory in Richland, WA, using a 12 Tesla (12 T) Bruker SolariX Fourier transform ion cyclotron resonance mass spectrometer (FTICR-MS) (Bruker, SolariX, Billerica, MA) with a custom direct infusion system (that performed two offline blanks between each sample) and an electrospray ionization (ESI) source. Data were acquired in negative mode with the needle voltage set to +4.0 kV and were collected from 150 m/z – 1000 m/z at 8 M. Three hundred scans were co-added for each sample and internally calibrated using OM homologous series separated by 14 Da (–CH2 groups). Mass measurement accuracy was generally within 1 ppm for singly charged ions across a broad m/z range (150 m/z - 1100 m/z). Raw spectra were converted to a list of m/z values using Bruker Data Analysis (version 5.0) by applying FTMS peak picker module with a signal-to-noise ratio (S/N) threshold set to 7 and absolute intensity threshold to the default value of 100. Chemical formulae were then assigned using Formularity19, an in-house software, following the Compound Identification Algorithm20,21,22. Criteria to assign chemical formulae included a S/N >7, and mass measurement error <0.5 ppm, taking into consideration the presence of C, H, O, N, S and P and excluding other elements19. Further processing of the data was done using the fticrrr R package23, including: (a) removing peaks <200 and >800 m/z, (b) removing peaks associated with 13C, and (c) blank correcting all spectra.
Soil and sediment analyses
Gravimetric water content
Gravimetric water content (GWC) was determined, calculated and reported as the dry moisture content24. Field moist soil (~5 g) was dried in the oven at 100 °C for 24 hours. Weight loss was then calculated using the following equation:
Dry weight basis is utilized herein to better indicate whether or not the soils were saturated24 across the broad spatial heterogeneity captured in EC1.
Bulk density
Bulk density was determined on intact cores collected in HYPROP rings, and calculated as:
Samples in the HYPROP rings were collected and maintained at field moisture, so the following conversion was applied to calculate the dry weight in the above equation:
Total carbon and nitrogen
Total carbon and nitrogen on a percent weight basis was determined via combustion and chromatographic separation using an ECS 8020 CHNS-O Elemental Analyzer (Orbit Technologies Pvt. Ltd.) equipped with a zero-blank electronic autosampler and thermal conductivity detector. Approximately 15 mg of freeze-dried, sieved, and homogenized soil were weighed into tin capsules. Reaction and reduction columns were packed according to operation manual specifications for C/N mode. For sample analysis, furnace temperatures were set to 980 °C for the reaction column, 650 °C for the reduction column, and 65 °C for the gas chromatograph. Carrier gas flow was held constant at ~110 ml/min. Standard reference sediments (SRM 1944; NY/NJ Waterway sediments) were run prior to each sample set, immediately following the calibration curve. We confirmed software peak detection, peak identification and integrations prior to exporting data. Calibration curves and final sample weight percentages were calculated in R Version 4.2.1 with the package EnvStats25.
Soil pH and conductivity
Soil pH and specific conductance were measured on freeze-dried and homogenized soils. Soil subsamples were shaken with deionized MilliQ water (1:10 weight:volume ratio) for 30 minutes and then analyzed using a Myron L 6PIIFCE pH and conductivity meter.
Data Records
Data (for complete list, see Table 2) are permanently deposited on the open access repository Environmental Systems Science Data Infrastructure for a Virtual Ecosystem (ESS-DIVE)26,27, accessible at https://doi.org/10.15485/196031310. Additional data types will be added to the ESS-DIVE data package as they are completed and will be version-controlled in the Change History section of README.pdf.
The structure of the data package is as follows:
Data Package Structure*
-
ec1_metadata_v1.zip
-
ec1_dd.csv: a file-level data descriptor file containing a list of every column present in the data files
-
ec1_flmd.csv: a file-level data descriptor file containing a list of every file name present in the data package
-
ec1_sample_catalog.csv: a file containing a list of all samples and their collection status or information about methodological inconsistencies
-
ec1_metadata_kitlevel.csv
-
ec1_metadata_collectionlevel.csv
-
ec1_data_collectionlevel.csv
-
ec1_igsn_metadata.csv
-
-
ec1_soil_v1.zip
-
ec1_sediment_v1.zip
-
ec1_water_v1.zip
-
ec1_processingscripts_v1.zip
*Please note the ESS-DIVE data package will include additional versions as we add new data types and the version number on the data package will reflect the latest version.
CSV file structure
-
[Campaign]_[Sample Type]_[Analyte]_[QC level].csv
-
Ex. ec1_soil_tctn_L2.csv
-
Ex. ec1_metadata_kitlevel_L2.csv
-
-
All .csv dataset files contain the following first three identifying columns:
-
campaign: coordinated sampling effort, Ex. EC1
-
kit_id: unique identifier for each collection of samples from a given site, Ex. K001
-
transect_location: position along the coastal TAI transect (Fig. 1), Ex. wetland
DAT file structure
-
-
[Kit_ID]_[Processing Step A]_[Processing Step…Z].dat
-
Ex. K004_DilCorr_IFE_RamNorm.dat
-
Ex. K013_DilCorr_Abs.dat
-
-
All .dat dataset files are organized by Kit_ID and in matrices.
Processing script structure
-
[Sample Type]_[Analyte].R
-
Ex. soil_tctn.R
-
Ex. water_cdom.R
-
-
All processing scripts follow a standardized structure outlined in template.R
Technical Validation
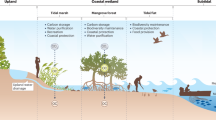
Technical validation steps were completed throughout the analysis process for each analyte (Fig. 2). Quality assurance of sample integrity was maintained from sample kit receiving, assuring that the quality of each sample was not compromised, by monitoring temperature and container quality upon kit arrival and stored properly for each analyte (Table 1). Instruments used to acquire EXCHANGE datasets were calibrated before each run and maintained using standard procedures for each instrument. Datasets were quality controlled following processing level designations (Table 3), inspired by the Ameriflux and Fluxnet programs28,29. For Level 1 (L1) datasets, flags are provided but are not applied. L1 datasets were screened for a secondary review and calculating the limit of detection ranges. Normal procedures for data quality were implemented, such as blank correction, etc, as appropriate. Analytical replicates are averaged, and outliers are also removed for L1 datasets. These datasets are archived on a Google Drive repository for additional data provenance and are accessible by the entire EXCHANGE consortium. For Level 2 (L2) datasets, all flags are applied to the L1 datasets, flagged data points removed, and data are summarized based on categorical variables (e.g., Transect Location, Kit ID). Datasets available on ESS-DIVE include L2 data for concentration-based datasets10.

Workflow of quality control procedures. Samples are received from the consortium, then processed at the Marine and Coastal Research Laboratory (PNNL–Sequim, WA) for analyses, which then were shared with the consortium and the public on ESS-DIVE.
We adopted the use of ESS-DIVE’s sample ID, file-level metadata, and CSV reporting formats30,31,32,33 to increase the usability of this data package and generate findable, accessible, interoperable and reusable (FAIR) data for the coastal science community33.
Usage Notes
This dataset follows Creative Commons Attribution 4.0 licensing, making all data freely available to use and distribute via the ESS-DIVE repository. Additional analyses are being performed on these sample sets, methods detailing these analyses can be found in the Supplementary Information. The data package10 will be updated periodically with such additional datasets, found at the same DOI, with version numbers of the data package indicating new datasets are available.
EXCHANGE is an open science, community-driven program. We encourage those that use this data for subsequent analyses to deposit their code in an open source repository, which aids in furthering our collective knowledge about coastal interfaces.
Code availability
All code necessary to reproduce our Level 2 datasets are written in the open source R Statistical Software34 version 4.2.2 and are publicly available in our ESS-DIVE repository accessible at https://doi.org/10.15485/196031310 in ec1_processingscripts_v1.zip. All scripts follow a standardized structure outlined in template.R and are named in the following format: [Sample Type]_[Analyte].R that correspond to their respective dataset name.
References
Ward, N. D. et al. Representing the function and sensitivity of coastal interfaces in Earth system models. Nat. Commun. 11, 2458 (2020).
Regier, P. et al. Biogeochemical control points of connectivity between a tidal creek and its floodplain. Limnol. Oceanogr. Lett. 6, 134–142 (2021).
Spivak, A. C., Sanderman, J., Bowen, J. L., Canuel, E. A. & Hopkinson, C. S. Global-change controls on soil-carbon accumulation and loss in coastal vegetated ecosystems. Nat. Geosci. 12, 685–692 (2019).
Hinson, A. L. et al. The spatial distribution of soil organic carbon in tidal wetland soils of the continental United States. Glob. Change Biol. 23, 5468–5480 (2017).
Baatz, R. et al. Steering operational synergies in terrestrial observation networks: Opportunity for advancing Earth system dynamics modelling. Earth Syst. Dyn. 9, 593–609 (2018).
Bernhardt, E. S. et al. Control Points in Ecosystems: Moving Beyond the Hot Spot Hot Moment Concept. Ecosystems 20, 665–682 (2017).
Trueblood, D. et al. Advancing Knowledge for Use in Coastal and Estuarine Management: Competitive Research in the National Estuarine Research Reserve System. Coast. Manag. 47, 337–346 (2019).
Lawson, R. Coordinating coastal wetlands monitoring in the North American Great Lakes. Aquat. Ecosyst. Health Manag. 7, 215–221 (2004).
Matuszeski, W. The Chesapeake Bay Program. Ekistics 62, 48 (1995).
Pennington, S. C. et al. EXCHANGE Campaign 1: A Community-Driven Baseline Characterization of Soils, Sediments, and Water Across Coastal Gradients. ESS-DIVE https://doi.org/10.15485/1960313 (2023).
Goldman, A. E., Emani, S. R., Pérez-Angel, L. C., Rodríguez-Ramos, J. A. & Stegen, J. C. Integrated, Coordinated, Open, and Networked (ICON) Science to Advance the Geosciences: Introduction and Synthesis of a Special Collection of Commentary Articles. Earth Space Sci. 9, (2022).
Dittmar, T., Koch, B., Hertkorn, N. & Kattner, G. A simple and efficient method for the solid-phase extraction of dissolved organic matter (SPE-DOM) from seawater. Limnol. Oceanogr. Methods 6, 230–235 (2008).
Rounds, S. A. Alkalinity and acid neutralizing capacity. US Geol. Surv. TWRI Book (2001).
Kopp, J. F. Methods for Chemical Analysis of Water and Wastes. (Environmental Monitoring and Support Laboratory, Office of Research and …, 1979).
Kelley, D., Richards, C. SCOR/IAPSO W. gsw: Gibbs Sea Water Functions. (2022).
Murphy, K. R., Stedmon, C. A., Graeber, D. & Bro, R. Fluorescence spectroscopy and multi-way techniques. PARAFAC. Anal. Methods 5, 6557–6566 (2013).
Ohno, T. Fluorescence inner-filtering correction for determining the humification index of dissolved organic matter. Environ. Sci. Technol. 36, 742–746 (2002).
Seidel, M. et al. Molecular-level changes of dissolved organic matter along the Amazon River-to-ocean continuum. Mar. Chem. 177, 218–231 (2015).
Tolić, N. et al. Formularity: software for automated formula assignment of natural and other organic matter from ultrahigh-resolution mass spectra. Anal. Chem. 89, 12659–12665 (2017).
Kujawinski, E. B. & Behn, M. D. Automated analysis of electrospray ionization Fourier transform ion cyclotron resonance mass spectra of natural organic matter. Anal. Chem. 78, 4363–4373 (2006).
Minor, E. C., Steinbring, C. J., Longnecker, K. & Kujawinski, E. B. Characterization of dissolved organic matter in Lake Superior and its watershed using ultrahigh resolution mass spectrometry. Org. Geochem. 43, 1–11 (2012).
Tfaily, M. M. et al. Sequential extraction protocol for organic matter from soils and sediments using high resolution mass spectrometry. Anal. Chim. Acta 972, 54–61 (2017).
Patel, K. F. kaizadp/fticrrr: FTICR-results-in-R. Zenodo https://doi.org/10.5281/zenodo.3893246 (2020).
Reddy, K. r., Clark, M. w., DeLaune, R. D. & Kongchum, M. Physicochemical Characterization of Wetland Soils. in Methods in Biogeochemistry of Wetlands 41–54. https://doi.org/10.2136/sssabookser10.c3 (John Wiley & Sons, Ltd, 2013).
Millard, S. P. & EnvStats-An, R. An R package for environmental statistics. (Springer, 2013).
Varadharajan, C. et al. Launching an accessible archive of environmental data. Eos 100 (2019).
Environmental System Science Data Infrastructure for a Virtual Ecosystem. ESS-DIVE https://data.ess-dive.lbl.gov/data.
Pastorello, G. et al. Observational Data Patterns for Time Series Data Quality Assessment. 2014 IEEE 10th International Conference on e-Science 1, 271–278 (2014).
Pastorello, G. et al. The FLUXNET2015 dataset and the ONEFlux processing pipeline for eddy covariance data. Sci. Data 7, 225 (2020).
Velliquette, T. et al. ESS-DIVE Reporting Format for Comma-separated Values (CSV) File Structure. (2021).
Velliquette, T. et al. ESS-DIVE Reporting Format for File-level Metadata. (2021).
Damerow, J. et al. Sample Identifiers and Metadata Reporting Format for Environmental Systems Science. (2020).
Crystal-Ornelas, R. et al. Enabling FAIR data in Earth and environmental science with community-centric (meta) data reporting formats. Sci. Data 9, 700 (2022).
R Core Team. R: A language and environment for statistical computing. (2022).
Acknowledgements
We would like to acknowledge the members of the EXCHANGE Consortium for their intellectual contributions and sampling efforts. Your commitment to community-driven science and collaboration have been instrumental in generating this new and extensive source of coastal ecosystem observational data. The EXCHANGE project is part of Coastal Observations, Mechanisms, and Predictions Across Systems and Scales - Field Measurements and Experiments (COMPASS-FME), a multi-institutional project supported by the U.S. Department of Energy, Office of Science, Biological and Environmental Research as part of the Environmental System Science Program. The Pacific Northwest National Laboratory is operated for DOE by Battelle Memorial Institute under contract DE-AC05-76RL01830. A portion of this research was performed at the Environmental Molecular Science Laboratory (EMSL) (grid.436923.9), a DOE Office of Science User Facility sponsored by the BER program. MRCAT/EnviroCAT beamline operations are supported by DOE and the member institutions. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Argonne National Laboratory is a U.S. Department of Energy laboratory managed by UChicago Argonne, LLC. The authors thank PNNL artist Nathan Johnson for preparing Figures 1 and 2 in this manuscript and Rosey Chu and Jason Toyoda for the collection of high-resolution mass spectrometry datasets.
Author information
Authors and Affiliations
Consortia
Contributions
Author contributions are detailed following the contributor roles taxonomy (CRediT) system by named author initials and/or the consortium name below. A full list of EXCHANGE consortium members is detailed alphabetically below. Conceptualization: A.N.M.P., N.D.W., A.M.L., V.B., S.C.P., O.O., K.M.K., EXCHANGE Consortium. Formal Analysis: A.N.M.P., S.C.P., K.F.P., P.R., O.O., K.K.H., J.A.R., L.S., D.J.D., Funding Acquisition: N.D.W., K.M.K., V.B., Investigation: A.N.M.P., S.C.P., K.P., O.O., M.B., L.S., E.J.O., D.J.D., C.G.N., M.I.B., K.M.K., K.K.H., N.C., EXCHANGE Consortium, Methodology: A.N.M.P., S.C.P., K.P., A.M.L., P.R., O.O., K.K.H., K.M.K., Project Administration: A.N.M.P., S.C.P., A.M.L., K.P., P.R., O.O., M.B., K.K.H., N.W., V.B., Resources: A.N.M.P., S.C.P., K.F.P., P.R., EXCHANGE Consortium, Software: S.C.P., P.R., J.A.R., Supervision: A.N.M.P., A.M.L., K.P., N.D.W., K.M.K., V.B., EXCHANGE Consortium, Validation: A.N.M.P., S.C.P., K.P., P.R., O.O., Visualization: A.N.M.P., S.C.P., A.M.L., Writing - original draft: A.N.M.P., S.C.P., K.F.P., A.M.L., P.R., O.O., L.S., K.M.K., K.K.H., Writing - review and editing: A.N.M.P., S.C.P., K.P., P.R., N.C., N.D.W., K.M.K., V.B.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Myers-Pigg, A.N., Pennington, S.C., Homolka, K.K. et al. Biogeochemistry of upland to wetland soils, sediments, and surface waters across Mid-Atlantic and Great Lakes coastal interfaces. Sci Data 10, 822 (2023). https://doi.org/10.1038/s41597-023-02548-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-023-02548-7
