
Abstract
Heteroptera (the true bugs), one of the most diverse lineages of insects, diversified in feeding strategies and living habitats, and thus become an ideal lineage for studies on adaptive evolution. Chinese water scorpion Ranatra chinensis (Heteroptera: Nepidae) is a predaceous bug living in lentic water systems, representing an ideal model for studying habitat transition and adaptation to water environment. However, genetic studies on this water bug remain limited. Here, we obtained a chromosome-level genome of R. chinensis using PacBio HiFi long reads and Hi-C sequencing reads. The total assembly size of genome is 867.89 Mb, with a scaffold N50 length of 26.48 Mb and the GC content of 39.50%. All contigs were assembled into 23 pseudo-chromosomes (N = 19 A + X1X2X3X4), and we predicted 18,424 protein-coding genes in this genome. This study will provide valuable genomic resources for future studies on the biology, water adaptation, and genome evolution of water bugs.
Similar content being viewed by others

Background & Summary
Biodiversity is the backbone of earth’s life support system, it plays a vital role in supporting and sustaining life on earth and maintaining the ecosystems functioning, it also underpins numerous essential benefits from nature that are vital for human well-being1,2,3. The ecosystem involved enormous organisms, raising an interesting question that how animals adapt into different habitats. The ability of flight and water adaptation, for example, enables animals gain novel niches and escape predators. For a number of animals, the molecular bases underlying the adaptive evolution have been uncovered. These animals include marine mammals4, Asian honeybee5, the water strider6 and the poultry shaft louse7. However, the adaptive mechanisms of the majority of extant species remain unknown.
Insects are a crucial component of biodiversity and play a vital role in maintenance of ecological balance, which are estimated to be as many as 5.5 million species on earth8. They make up around 80% of animals and around half of all living species, and occupy nearly every terrestrial habitat on the planet8,9. The true bugs (Hemiptera: Heteroptera) is one of the most diverse lineages of insects and the most diversified lineage among hemimetabola, which possesses a diversity of feeding strategies, ranging from predation on other arthropods and hematophagy on vertebrates, to mycetophagy and phytophagy. Hemipterans also vary in living habitats, including various terrestrial, aquatic and even marine habitats10,11. The enormous diversity makes Hemiptera an ideal lineage for exploration of adaptive evolution. In previous studies, the common ancestor of Heteroptera was considered as terrestrial and experienced multiple independent evolutionary events on habitat transitions, from terrestrial to water surface, aquatic and other habitation, and from aquatic to shoreline11,12. The infraorder Nepomorpha, specifically, containing many species living in water, is helpful for understanding on water adaptation and habitats transition. Exploring the water adaptation of water bugs can pave the way for the following studies related to living habitat adaptation. However, the lack of chromosome-level genomes prevents us from a deep genomic analysis of aquatic true bugs and also impedes the discovery of the adaptive mechanism.
The family Nepidae (Heteroptera: Nepomorpha), or water scorpions, can be recognized by the characteristics of the antennae hidden under the head and the long tail-like siphon (the respiratory siphon) on the rear end of their body that cannot be retracted into the apex of the abdomen, which is unique within the aquatic insects13,14,15. Normally, they can be found in ponds, lakes, marshes and riversides and often move far from its aquatic habitat at night16. As predatory insects, they usually reside in the shade of plants in water or between stones, waiting for its preys, including small fish and aquatic insects and other invertebrates in water16. This means, Nepidae have evolved multiple derived traits compared to ancestors and are also potential natural enemies of health pests for biocontrol, because they are found to be able to control mosquito population by preying the larvae17,18. Ranatra chinensis, also known as Chinese water scorpion, is a representative species of family Nepidae with a long caudally breathing tube. Its body size ranges from 41 mm to 48 mm, and the length of its respiratory siphon can be equal to that of torso18. Ranatra chinensis is widely distributed in China, from the northernmost Heilongjiang Province to southernmost Guangdong Province, making it a relatively accessible specimen for scientific studies.
There is no chromosome-level genome available within family Nepidae yet. Analysis on R. chinensis genome will benefit our interpretation about the molecular mechanisms of water bugs’ habitat transition and adaptative evolution. With the development of sequencing technologies and bioinformatic tools, obtaining a high-quality reference genome becomes feasible for most organisms, enabling us to investigate the genome evolution and the underlying molecular mechanisms. In this study, we assembled and annotated the chromosome-level genome of R. chinensis, combining of PacBio long-read, Illumina short-read sequencing, and chromosome conformation capture (Hi-C) technologies. Our data will provide genomic resources for future exploration on the biology and evolution of water bugs, and will facilitate the understanding of habitat transition and water adaptation.
Methods
Sample preparation
The R. chinensis specimen were collected from Xichuan County, Henan Province, China (33.24° N, 111.02° E), and were put into dry ice for transportation. All samples were stored at −80°C until further usage. The female adult was used for genomic DNA extraction based on the CTAB method, and extracted DNA were purified using a Blood and Cell Culture DNA Midi Kit (QIAGEN, Germany). The DNA degradation and contamination of the extracted DNA was monitored on 1% agarose gels. The purity of DNA samples was then detected using NanoDrop™. One UV-Vis spectrophotometer (Thermo Fisher Scientific, USA) and the Qubit® 4.0 Fluorometer (Invitrogen, USA) were used to measure the DNA concentration.
Genomic DNA sequencing
Illumina short read library was firstly constructed with an insertion size of about 350 bp, and then sequenced using the Illumina Novaseq6000 to generate 150 bp paired-end short reads. We obtained 59.89 Gb raw data of Illumina short reads and finally got 57.55 Gb clean data after removing adapters and low-quality short reads using Fastp version 0.21.019 with default parameters (Table 1).
For long-read sequencing, a PacBio HiFi-read library with insertion sizes of 15 kb was generated, and sequenced the long DNA fragments using a SMRT cell on PacBio Sequel II sequencing platform (Pacific Biosciences, Menlo Park, USA). A total of 60.41 Gb clean data were obtained from the raw long reads generated using Circular Consensus Sequencing (CCS) model (Table 1).
A single female adult was used for chromosome conformation capture (Hi-C) sequencing and the library was prepared according to the standard protocol described by Belton with minor modifications20. The sample was cut into pieces and mixed with 2% formaldehyde solution for cross-linking, and then treated with New England Biolabs (NEB) buffer to digest nuclei. Biotinylated nucleotides were used to fill the cohesive ends and purified DNA was sheared to fragments of 350 bp in length after ligation. DNA purification was achieved using QIAamp DNA Mini Kits (Qiagen). The final generated Hi-C library was sequenced on Illumina Novoseq6000 platform with paired-end 150 bp. The sequencing yields 59.18 Gb raw data and 57.87 Gb clean data obtained after applying the same filter criteria for short reads (Table 1).
Transcriptome sequencing
Total RNA was extracted from two adults (one female and one male) with the TRIzol reagent (Thermo Fisher Scientific, USA) for transcriptome sequencing. The construction of a paired-end library was obtained by using the TruSeq RNA Library Preparation Kit (Illumina, USA). The transcriptome sequencing was finished on an Illumina Novoseq6000 platform, resulting in a total of 4.86 Gb RNA-seq clean data (Table 1).
Genome size estimation
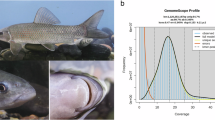
Genomic characteristics including genome size, heterozygosity, and duplication were estimated using 57.55 Gb clean Illumina short-reads. The distribution of k-mer copy number was calculated to perform this estimation in JELLYFISH version 2.1.321. Genome size and genome heterozygosity were estimated based on 17-mer depth analysis in GenomeScope version 2.022 with default parameters, and the results were 605.54 Mb and 2.27%, respectively (Fig. 1).

The K-mer distribution of Illumina paired-end reads using GenomeScope version 2.0 based on a k value of 17. The K-mer distributions showed double peaks: the first peak with a coverage of ~40 indicates genome duplication, and the second peak with a coverage of ~60 represents a genome size peak.
Chromosome-level genome assembly
The initial de novo assembly of R. chinensis genome was performed based on PacBio sequence data using Hifiasm version 0.13 with default parameters23. After assembly, the genome was then polished by the Purge_Dups pipeline24 to remove alternative haplotype and redundant fragments from the genome. Then, the subsequent polishing was performed using Illumina sequencing data to enhance the quality of the contigs. Finally, an 867.89 Mb contig-level genome assembly of R. chinensis was obtained based on PacBio sequencing data, containing 689 contigs with contig N50 and N90 sizes of 26.48 Mb and 3.80 Mb, respectively, and the GC content of 39.50% (Table 2).
The high-quality chromosome-scale genome was generated using a scaffolding pipeline based on Durand25. Initially, BWA-MEM version 0.7.1726 with the parameters: ‘mem -SP5M’ was used for mapping Hi-C data to the contig assembly genome. The DpnII sites were generated using the ‘generate_site_positions.py’ script in Juicer version 1.525. Subsequently, contigs were assembled into the chromosome-level scaffolds using the 3D-DNA pipeline with the parameter “-r 2”27. After the confirmation by Hi-C contact maps, chromosome interaction matrix was manually adjusted and corrected using Juicebox version 1.11.0828. Ultimately, we anchored and generated 23 pseudo-chromosomes, and the final chromosome-level genome assembly of R. chinensis was obtained with a scaffold N50 of 29.80 Mb (Fig. 2; Table 2).

The Circos atlas of the Ranatra chinensis chromosome-level genome. Tracks represent (a) the distribution of chromosome karyotypes, (b) gene density, (c) transposable element content, (d) DNA transposon and (e) GC density. Densities were calculated in 100-kb windows. Chr8, Chr21, Chr22 and Chr23 are predicted to be the sex chromosomes and the remaining are autosomes.
Synteny analysis and the determination of sex chromosome in R. chinensis
Based on JCVI v1.1.17 with default parameters29, we performed synteny analysis to confirm the sex chromosome in R. chinensis using public chromosome-level genomes whose sex chromosomes have been verified, including Rhynocoris fuscipes (Hemiptera: Reduviidae)30, Triatoma rubrofasciata (Hemiptera: Reduviidae)31 and Riptortus pedestris (Hemiptera: Alydidae)32. The result showed that the Chr8, Chr21, Chr22 and Chr23 of R. chinensis exhibited high homology with Chr12 and Chr13 of T. rubrofasciata, Chr13, Chr14 and Chr15 of R. fuscipes, and the ChrX of R. pedestris (Fig. S1). This result indicates that R. chinensis has the same sex chromosome system as that of Nepa cinerea (Heteroptera: Nepidae, N = 14 A + X1X2X3X4) and Ranatra linearis (Heteroptera: Nepidae, N = 19 A + X1X2X3X4) reported in previous study33, and Chr8, Chr21, Chr22 and Chr23 correspond to the X1, X2, X3, and X4 chromosomes.
Prediction of repetitive elements
Repeat sequences of R. chinensis genome were detected in Extensive de novo TE Annotator (EDTA) version 1.9.434. LTR retrotransposons were determined in LTR FINDER version 1.0735, LTRharvest36, and LTR retriever version 2.9.037 with default parameters. DNA transposons were classified utilizing TIR Learner38 and HelitronScanner39 with default parameters. RepeatMasker version 4.0.7 (parameters: -gff -xsmall -no_is)40 and RepeatProteinMask version 4.0.7 (parameters: -engine wublast) were used to find the interspersed repeats against the RepBase database41 (http://www.girinst.org/repbase). In addition, Tandem Repeats Finder version 4.0942 was used to classify tandem repeats with parameters ‘2 7 7 80 10 50 500 -f -d -m’ based on the de novo prediction. RepeatModeler version 2.0.443 (parameters: ‘-engine ncbi -pa 4’) was utilized to construct a repetitive sequence library and RepeatMasker version 4.0.740 was used for annotation of the repeat element against this repeat library. In the genomic sequences, a total of 391.73 Mb (45.13%) repetitive elements were identified, mainly including 22.96% retrotransposon, 3.35% DNA transposons and 11.26% tandem repeat (Table 3). Retrotransposons include LTR, SINE, and LINE; and LTR is further classified in to Copia, Gypsy, and other LTR.
Protein-coding gene prediction and functional annotation
Protein-coding genes (PCGs) within the genome were predicted by a combined method of homology-based prediction, ab initio prediction and transcriptome-based prediction. HISAT2 version 2.2.144 was utilized to map RNA-seq short data to the genome with the parameter ‘-k 2’. Then the StringTie version 2.4.045 was used to assemble the mapped reads into transcripts with default parameters. For the homology-based prediction, the protein sequences of eight representative insect species were downloaded from the NCBI GenBank database (Table S1). Homologous proteins were aligned against R. chinensis genome using Exonerate version 2.4.0 with default parameters to train the gene sets. Additionally, the bam2hints program (parameter: -intronsonly) in AUGUSTUS version 3.2.346 was employed to transfer the sorted and mapped bam file of RNA-seq data into a hints file. To predict coding genes from the assembled genome, AUGUSTUS version 3.2.346 with default parameters was performed for prediction, in which the combination of trained gene sets and hint files was the input. In the end, MAKER version 2.31.1047 was utilized to merge and generate a consensus high-confidence gene set on the basis of homology-based, de novo-derived and transcript genes. We predicted a total of 18,424 genes in R. chinensis genome with an average gene length of 5,900.48 bp (Table 4). The average length of coding sequences (CDS) and protein sequence were 1,189.79 bp and 396.60 AA, respectively (Table 4). The above statistics on sex chromosomes and autosomes were provided in Table S2.
The predicted genes were functionally annotated using multiple methods, includeincludeing eggnog-mapper48 (parameter: -m diamond–tax_scope auto–go_evidence experimental–target_orthologs all–seed_ortholog_evalue 0.001–seed_ortholog_score 60–query-cover 20–subject-cover 0 –override), InterProscan version 5.049 (parameter: -iprlookup -goterms -appl Pfam -f TSV), BLAST version 2.2.2850 (parameter: -evalue 1e-5), and HMMER version 3.3.251 (parameter: –noali–cut_ga Pfam-A.hmm). All these approaches were performed to search against several public databases: Gene Ontology (GO), Clusters of Orthologous Groups of Proteins (COG), Kyoto Encyclopedia of Genes and Genomes (KEGG), NCBI non-redundant protein (Nr), Swiss-Prot, and Pfam. Overall, 16,262 genes were functionally annotated with at least one public database (Table 5).
Data Records
Genomic Illumina short-reads data were deposited at the NCBI Sequence Read Archive database under accession number SRR2878529252. Genomic PacBio HiFi sequencing data were deposited at the NCBI Sequence Read Archive database under accession number SRR2878938053. RNA-seq data was deposited at the NCBI Sequence Read Archive database under accession number SRR2878888054. The Hi-C sequencing data were deposited at the NCBI Sequence Read Archive database under accession number SRR2878753855.
The final chromosome assembly was submitted to GenBank at NCBI under accession number JBFDAA00000000056. The genome sequence and raw reads have been deposited in GenBank and Sequence Read Archive at NCBI under BioProject PRJNA110371857.
Technical Validation
To assess the accuracy of the final genome assembly, we mapped the Illumina short-reads to the R. chinensis genome with BWA-MEM version 0.7.1726, and the result showed 97.08% of short reads were successfully mapped to the genome. Benchmarking Universal Single-Copy Orthologs (BUSCO version 3.0.2)58 was used to evaluate the genome completeness based on the insecta_odb10 database, revealing the completeness was 95.7%. Among 1,309 orthologous, 1,299 genes were classified as complete single-copy genes and 10 genes were complete duplicated genes, eight genes were fragmented and 48 genes were missing (Table 6).
Code availability
The bioinformatic analyses were performed using the manuals and protocols by the software developers, with the manually adjusted parameters clearly described in the Methods. No custom script or code was used in this study.
References
Cardinale, B. et al. Biodiversity loss and its impact on humanity. Nature 486, 59–67, https://doi.org/10.1038/nature11148 (2012).
Brauman, K. A. et al. Global trends in nature’s contributions to people. Proc. Natl. Acad. Sci. USA 117, 32799–32805, https://doi.org/10.1073/pnas.2010473117 (2020).
Kim, H. J. et al. Understanding the role of biodiversity in the climate, food, water, energy, transport and health nexus in Europe. Sci. Total Environ. 925, 171692, https://doi.org/10.1016/j.scitotenv.2024.171692 (2024).
Yuan, Y. et al. Comparative genomics provides insights into the aquatic adaptations of mammals. Proc. Natl. Acad. Sci. USA 118, e2106080118, https://doi.org/10.1073/pnas.2106080118 (2021).
Ji, Y. et al. Gene reuse facilitates rapid radiation and independent adaptation to diverse habitats in the Asian honeybee. Sci. Adv. 6, eabd3590 https://www.science.org/doi/10.1126/sciadv.abd3590 (2020).
Santos, M., Le Bouquin, A., Crumière, A. & Khila, A. Taxon-restricted genes at the origin of a novel trait allowing access to a new environment. Science 358, 386–390, https://www.science.org/doi/10.1126/science.aan2748 (2017).
Xu, Y. et al. Chromosome-level genome of the poultry shaft louse Menopon gallinae provides insight into the host-switching and adaptive evolution of parasitic lice. GigaScience 13, giae004, https://doi.org/10.1093/gigascience/giae004 (2024).
Stork, N. E. How many species of insects and other terrestrial arthropods are there on earth? Annu. Rev. Entomol. 63, 31–45, https://doi.org/10.1146/annurev-ento-020117-043348 (2018).
Slade, E. M. & Ong, X. R. The future of tropical insect diversity: strategies to fill data and knowledge gaps. Curr. Opin. Insect Sci. 58, 101063, https://doi.org/10.1016/j.cois.2023.101063 (2023).
Schuh, R. T. & Slater, J. A. True bugs of the world (Hemiptera: Heteroptera) classification and natural history. Cornell University Press, Ithaca, NY. (1995).
Weirauch, C., Schuh, R. T., Cassis, G. & Wheeler, W. C. Revisiting habitat and lifestyle transitions in Heteroptera (Insecta: Hemiptera): insights from a combined morphological and molecular phylogeny. Cladistics 35, 67–105, https://doi.org/10.1111/cla.12233 (2019).
Li, H. et al. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. R. Soc. B 284, 20171223, https://doi.org/10.1098/rspb.2017.1223 (2017).
Chen, P., Nieser, N. & Ho, J. Review of Chinese Ranatrinae (Hemiptera: Nepidae), with descriptions of four new species of Ranatra Fabricius. Tijd. Entomol. 147, 81–102, https://api.semanticscholar.org/CorpusID:84596642 (2004).
Chen, P., Nieser, N. & Zettel, H. The aquatic and semi-aquatic bugs (Heteroptera: Nepomorpha & Gerromorpha) of Malesia. Fauna Malesiana Handbooks 5. Leiden and Boston, Brill (546 pp), https://api.semanticscholar.org/CorpusID:82862404 (2005).
Polhemus, D. A. & Polhemus, J. T. Guide to the aquatic Heteroptera of Singapore and Peninsular Malaysia. X. Infraorder Nepomorpha – Families Belostomatidae and Nepidae. Raffles Bull. Zool. 61, 25–45, https://api.semanticscholar.org/CorpusID:87760711 (2013).
Ueno, M. Hemiptera. In: Freshwater Biology of Japan. (Edited by UCno M.), Hokuryukan Pub. Co. Ltd, Tokyo. pp. 567–575, (1973).
Shaalan, E. A. & Canyon, D. V. Aquatic insect predators and mosquito control. Trop. Biomed. 26, 223–261, https://pubmed.ncbi.nlm.nih.gov/20237438/ (2009).
Cui, J. & Cai, W. Taxonomic note on Ranatra Fabricius from Henan of China. Journal of Henan Agricultural Sciences 44, 87–90, https://www.hnnykx.org.cn/EN/Y2015/V44/I2/87 (2015).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Belton, J. M. et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Cheng, H. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–5, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488, https://www.science.org/doi/10.1126/science.1153917 (2008).
Ma, L. et al. Comparative genomic analyses on assassin bug Rhynocoris fuscipes (Hemiptera: Reduviidae) reveal genetic bases governing the diet-shift. iScience 27, 110411, https://doi.org/10.1016/j.isci.2024.110411 (2024).
Liu, Q. et al. A chromosomal-level genome assembly for the insect vector for Chagas disease, Triatoma rubrofasciata. GigaScience 8, giz089, https://doi.org/10.1093/gigascience/giz089 (2019).
Huang, H. J. et al. Chromosome-level genome assembly of the bean bug Riptortus pedestris. Mol. Ecol. Resour. 21, 2423–2436, https://doi.org/10.1111/1755-0998.13434 (2021).
Angus, R. B., Jeangirard, C., Stoianova, D., Grozeva, S. & Kuznetsova, V. G. A chromosomal analysis of Nepa cinerea Linnaeus, 1758 and Ranatra linearis (Linnaeus, 1758) (Heteroptera, Nepidae). Comp. Cytogenet. 11, 641–657, https://pubmed.ncbi.nlm.nih.gov/29114353/ (2017).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 1–18, https://doi.org/10.1186/s13059-019-1905-y (2019).
Ou, S. & Jiang, N. LTR_FINDER_parallel: parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mobile DNA 10, 48–48, https://doi.org/10.1186/s13100-019-0193-0 (2019).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 9, 1–14, https://doi.org/10.1186/1471-2105-9-18 (2008).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422, https://doi.org/10.1104/pp.17.01310 (2017).
Su, W., Gu, X. & Peterson, T. TIR-Learner, a new ensemble method for TIR transposable element annotation, provides evidence for abundant new transposable elements in the maize genome. Mol. Plant 12, 447–460, https://doi.org/10.1016/j.molp.2019.02.008 (2019).
Xiong, W., He, L., Lai, J., Dooner, H. K. & Du, C. HelitronScanner uncovers a large overlooked cache of Helitron transposons in many plant genomes. Proc. Natl. Acad. Sci. USA 111, 10263–10268, https://doi.org/10.1073/pnas.141006811 (2014).
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 5, 1–14, https://doi.org/10.1002/0471250953.bi0410s25 (2004).
Jurka, J. et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467, https://doi.org/10.1186/s13100-015-0041-9 (2005).
Benso, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 1–13, https://doi.org/10.1186/s13059-019-1910-1 (2019).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, 435–439, https://doi.org/10.1093/nar/gkl200 (2006).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196, http://www.genome.org/cgi/doi/10.1101/gr.6743907 (2008).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829, https://doi.org/10.1093/molbev/msab293 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinform. 10, 421–429, https://doi.org/10.1186/1471-2105-10-421 (2009).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, 29–37, https://doi.org/10.1093/nar/gkr367 (2011).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR28785292 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR28789380 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR28788880 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR28787538 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBFDAA000000000 (2024).
NCBI BioProject https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1103718 (2024).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 32120103006, 31922012) and the 2115 Talent Development Program of China Agricultural University. We thank Shuxian Chen for the help in statistics.
Author information
Authors and Affiliations
Contributions
Y.D. and W.C. conceived the project. L.M., X.L., Y.W. and T.X. collected samples and extracted genomic DNA. L.M., X.L. and Y.W. performed data analysis and wrote the manuscript. L.T., F.S., T.X. and H.L. contributed to data analyses. All authors contributed to revising the manuscript. All authors have read and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, X., Ma, L., Tian, L. et al. Chromosome-level genome assembly of Chinese water scorpion Ranatra chinensis (Heteroptera: Nepidae). Sci Data 11, 1016 (2024). https://doi.org/10.1038/s41597-024-03856-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-03856-2
