
Abstract
The retina plays a crucial role in processing and decoding visual information, both in normal development and during myopia progression. Recent advancements have introduced a library-independent approach for data-independent acquisition (DIA) analyses. This study demonstrates deep proteome identification and quantification in individual mice retinas during myopia development, with an average of 6,263 ± 86 unique protein groups. We anticipate that the use of a predicted retinal-specific spectral library combined with the robust quantification achieved within this dataset will contribute to a better understanding of the proteome complexity. Furthermore, a comprehensive mice retinal-specific spectral library was generated, encompassing a total identification of 9,401 protein groups, 70,041 peptides, 95,339 precursors, and 761,868 transitions acquired using SWATH-MS acquisition on a ZenoTOF 7600 mass spectrometer. This dataset surpasses the spectral library generated through high-pH reversed-phase fractionation by data-dependent acquisition (DDA). The data is available via ProteomeXchange with the identifier PXD046983. It will also serve as an indispensable reference for investigations in myopia research and other retinal or neurological diseases.
Similar content being viewed by others

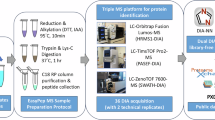

Background & Summary
Myopia (shortsightedness) has been increasing globally in recent decades1. Its serverity can lead to high myopia (≤−6.0 diopter) on the basis of uncorrected myopia that can significantly impact visual acuity to the extent of meeting the threshold for blindness2. The neural retina is a light-sensitive tissue composed of 10 morphological layers connected by neuronal synapses. The complexity of the retinal proteome is evident from the presence of six major cell types characterized by marker expressions in rods (Pde6a), cones (Pde6c), bipolar cells (Vsx2), amacrine cells (Pax6), Müller glia cells (Vim), horizontal cells (Onecut1), and retinal ganglion cells (Slc17a6), as well as 38 retinal clusters identified by single-cell transcriptomics3. However, the lack of retinal cell lines comparable to the neural retina has hindered research in myopia and other retinal or neurological diseases, with only limited ocular cell lines available for retinal pigment epithelium (RPE)4,5, arising retinal pigment epithelia cell line ARPE-19 and D407, retinal ganglion cell line RGC-56,7, and cone photoreceptor cell8 line 661 W. Therefore, understanding the retinal profile in normal and myopia development is crucial for investigating retinal proteins in myopia9 and retinal diseases10,11,12. Additionally, the retina has emerged as a potential gateway for screening and monitoring neurological diseases such as Alzheimer’s disease13,14,15,16, schizophrenia17,18, and Parkinson’s disease19,20.
Sequential window acquisition of all theoretical mass spectra (SWATH-MS) is a specific variant of data-independent acquisition (DIA) that enables consistent, reproducible quantitative proteomics results21,22. Traditional DIA workflow required tissue-specific spectral libraries with extensive proteome coverage generated through fractionation techniques such as high-pH reversed-phase peptide fractionation23, strong cation-exchange (SCX) chromatography24, or gas-phase fractionation25. In a precious study, a retinal-specific spectral library was generated using high-pH reserved-phase peptide fractionation, consisting of 5,609 proteins and 50,776 peptide sequences, acquired through data-dependent acquisition (DDA) on the TripleTOF 6600 mass spectrometer (Q-TOF, Sciex)26. However, recent advancements have introduced a library-free approach27,28,29, for DIA analysis facilitated by DIA-NN30. DIA-NN utilized neural networks to search the in-silico protein digestion of the reference proteome, which is an approach previously introduced27,29. This apporach eliminates the prerequisite spectral library and enables flexibility in analyzing DIA data from different mass spectrometry vendors and software tools. Recent studies have demonstrated the superior performance of DIA-NN compared to other software, supporting various raw data, and spectral library formats30,31
This study demonstrates deep proteome identification and quantification without fractionation during myopia development, resulting in an average of 6,263 ± 86 unique protein groups, surpassing the reported spectral library prepared through fractionation with quantitative analysis in individual mice retinas. Furthermore, a comprehensive mice retinal-specific spectral library was generated using DIA-NN, comprising a total identification of 9,401 protein groups, 70,041 peptides, 95,339 precursors, and 761,868 transitions. This retinal-specific spectral library is comparable to spectral libraries derived from multiple organs with 11,000 proteins that lack of eye tissues in mice32 and guinea pig33. Additionally, it is also comparable to the spectral libraries generated from multiple rabbit eye tissues prepared through high-pH reserved-phase fractionation34. This dataset demonstrates proprietary protocols for protein analysis of murine retina utilizing simple, ultrafast sample preparation with S-Trap35, robust quantification via SWATH-MS, and a library-free approach with the MaxLFQ36 algorithm using DIA-NN. The in-depth retinal-specific spectral library will significantly contribute to a better understanding of the proteome complexity of the neural retina. Moreover, it will serve as an indispensable reference for investigating retinal proteomes in myopia research and other retinal or neurological diseases.
Methods
Animals
Black C57BL/6 J wild-type mice were obtained from The Jackson Laboratory (Farmington, CT, USA). Mice were maintained as in-house breeding colonies at the centralized animal facility of The Hong Kong Polytechnic University. Animals were housed in standard mouse cages (Sealsafe Plus GM500, Tecniplast, Varese, Italy) at a temperature of 25 °C with a 12:12 hour light/dark cycle in a room with a light intensity of 150 lux. Food and water were provided ad libitum. Mice were weaned on postnatal day 21 and housed until postnatal day 35. The researchers involved in this study were licensed by the Department of Health, HKSAR government. All procedures performed in this study received ethics approval from the Animal Subjects Ethics Sub-Committee (ASESC) of The Hong Kong Polytechnic University and complied with the Association of Research in Vision and Ophthalmology (ARVO) statement for the use of animals in ophthalmology and vision research.
Lens-induced myopia in C57BL6/J mice
For all procedures in mice, including fixation of the spectacle system, refractive error, and ocular biometric measurements, anesthesia was administered. Briefly, the pupil of each eye was dilated with mydrin-P ophthalmic solution, which contained 0.5% tropicamide and 0.5% phenylephrine HCl for 15 minutes. The mouse was then sedated through an intraperitoneal injection of a mixture of ketamine (70 mg/kg) and xylazine (10 mg/kg). The customized spectacle system, previously described in a publication, was assembled onto the frame in advance37. A randomized eye, unilateral lens-induced myopia (LIM) treatment was applied in the study from postnatal day 21 to 35. Ocular biometrics were measured at P21, P28, and P35 using an infrared photorefractor and SD-OCT.
Ocular biometric measurements in mice
The eccentric infrared photorefractor was purchased from the manufacturer (Steinbeiss-Transfer Centre for Biomedical Optics, Tuebingen, Germany) and used according to the user manual38. Refractive errors were acquired by aligning to the Purkinje image from the corneal reflection, known as P1, using software-controlled gaze control in the x- and y-axes, with a tolerance of 5 or less. Additionally, each measurement was carefully adjusted to be as close as possible to the x- and y-axes near zero, collecting 99 measurements per eye. The measurement was represented as the mean value in diopter ± SD and repeated in triplicate. The ocular dimension was measured in radial volume mode, with duplicate measurement (A-scans = 1000 lines, B-scans = 6, 32 frames, 80 lines of inactive A-scans, 0.4 mm diameter). The length of each component was represented as the mean value in micrometers. Spectral domain-optical coherence tomography (SD-OCT, Envisu R4310, Leica, Germany) equipped with a 50° probe for mice was used to measure the segmented ocular dimensions. After each SD-OCT measurement, the mice were reset to the default position, and duplicate measurements were acquired. The ocular segmentation was manually analyzed using a digital caliper in OCT data analysis software (InVivoVue, ver. 2.4, Leica). Axial length (AL) was represented as the depth from corneal surface to the retinal pigment epithelium layer.
Tissue homogenization and protein extraction
Retinas (n = 22) were individually homogenized using a liquid nitrogen cooled tissue homogenizer (Precellys Evolution, Bertin Instruments. The homogenization was performed with 250 μL of lysis buffer containing 5% sodium dodecyl sulfate (SDS) and 50 mM triethylammonium bicarbonate (TEAB) for mouse retina, while 350 μL of lysis buffer was optimized for chick retina. The sample was homogenized at 7,000 rpm for 30 s × 4 cycles with 20 s intervals, at 4 °C in a homogenization tube (CKMix, Bertin Instruments, France). After homogenization, the sample was briefly centrifuged, and the supernatant was transferred to a new 1.5 mL Eppendorf tube. It was then centrifuged at 15,000 rpm for 30 min at 4 °C. The protein concentration was determined using the bicinchoninic acid assay (Pierce Rapid Gold BCA Protein Assay, A53225, Thermo Fisher Scientific).
Mass spectrometry sample preparation
The protocol was adopted from a previous publication and followed the manufacturer’s procedure26,35. Suspension trap columns (S-Trap micro, Protifi, USA) were purchased for the experiment. A total of 50 μg of protein was reduced with a final concentration of 20 mM dithiothreitol (DTT) at 95 °C for 10 min. After cooling to room temperature, the protein was alkylated with a final concentration of 40 mM iodoacetamide (IAA) at room temperature in the dark for 10 min. The SDS lysate was acidified with a final concentration of 1.2% aqueous phosphoric acid. The solution was diluted using six volumes of protein binding buffer (90% methanol, 0.1 M TEAB, pH 7.5). The mixture was loaded onto an S-Trap spin column and centrifuged at 4,000 g for 20 s. The captured protein was washed with 150 μL of protein binding buffer and centrifuged at 4,000 g for 20 s. This washing step was repeated three times. Protein digestion was performed by adding 20 μL of digestion buffer (50 mM TEAB) containing trypsin (V5111, Promega, USA) at a ratio of 1:25 (w/w, trypsin: protein) to the filter, and the mixture was digested at 47 °C for 1 hour. Peptides were eluted in three sequential steps: 40 μL of 50 mM TEAB, 40 μL of 0.2% aqueous formic acid (FA), and 35 μL of 0.2%FA in 50% acetonitrile. The three eluted peptide solutions were pooled and lyophilized using vacuum centrifugation at 4 °C (CentriVap, Labconco, USA). The peptides were then resuspended in 20 μL of 0.1% FA. The peptide concentration was measured and normalized to 0.5 μg/μL with 0.1% FA solution using a colorimetric peptide assay (Cat. No. 23275, Thermo Fisher Scientific) for mass spectrometric analysis.
Data-independent acquisition of the mouse retina proteome
Peptides (1 μg) were acquired in duplicate using SWATH-MS mode. The MS1 survey scans were acquired from 350 to 1800 Da with an accumulation time of 50 ms. Precursors were fragmented in CID mode, measured with 100 variable windows ranging from 350 to 1800 Da with a 25 ms accumulation time and rolling collision energy. The scheme of 100 isolation windows was determined based on precursor frequencies in DDA acquisition of the mouse retina sample using the SWATH variable window calculator (Ver. 1.2, Sciex). (Supplementary Table S1) Peptides were loaded with isocratic loading buffer (2% ACN, 0.1% FA) onto a reversed-phase chromatography trap column packed with 5 μm, 180 μm × 20 mm (nanoEase M/Z Symmetry C18, Waters) at a flow rate of 3 μL/min for 15 min. Chromatographic separation was performed using a C18 reversed-phase analytical column with 1.8 μm, 75 μm × 200 mm (nanoEase M/Z HSS C18 T3, Waters) at a flow rate of 0.3 μL/min. The following gradient was used: 0–0.5 min: 5% B (ACN, 0.1% FA), 0.5–90 min: 10% B, 90–120 min: 20% B, 120–130 min: 28% B, 130–135 min: 45% B, 135–141 min: 80% B, and finally equilibration between 141–155 min with 5% B using mobile phase A (0.1%FA, deionized water). The nano-microscale liquid chromatography was performed using the ACQUITY UPLC M-Class system (Waters). The column outlet was connected to an OptiFlow Turbo V ion source equipped with a NanoSpray probe (Cat. No. 5029431, Sciex) on a ZenoTOF 7600 mass spectrometer from Sciex (Concord, Ontario, Canada).
Quantitative analysis and the generation of retinal-specific spectral library
The raw data files in.wiff format were analyzed using DIA-NN (ver. 1.8.1) in library-free mode with default settings. The analysis was performed in “robust LC (high precision)” with precursor m/z values ranging from 300 to 1800, precursor charges between 1 and 4, MS1 and MS2 accuracy set to 20 ppm and 10 scan windows. The match-between run (MBR) option was enabled30. Protein quantification was carrired out using the MaxLFQ algorithm implemented in the software. Protein identities were assigned by searching against the in-silico tryptic digestion using the UniProt Mus Musculus reference database (UP000000589, 17173 reviewed proteins, Oct 2023).
Data Records
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE39 partner repository with the dataset identifier PXD04698340. Each run is identified by its date, unique reference number, eyes (OS/OD), treatment group (LIM/CTL), and technical replication (T1/T2). This structured file name can be easily expanded into columns using the delimiter. Each uploaded raw file consists of 4 native types known as.wiff,.wiff2,.wiff.scan, and.timeseries.data from the Sciex mass spectrometer. Additionally, each uploaded raw file is accompanied by a.wiff.quant file generated by the software DIA-NN. There are three.tsv file-based outputs generated from the dataset. The library.tsv file contains the mass spectrometry-based spectral library. The report.pg_matrix.tsv file contains all protein groups found by searching the associated raw file. The report.pr_matrix.tsv file contains aggregated evidence for each identified peptide. The matrix.tsv file contains a matrix table that expands both horizontally by sample and vertically by protein group or peptide. The number of rows and columns in the matrix is determined by the number of unique values in the specified fields, represented as intensity computed by the MaxLFQ algorithm. Users can also refer to the developer manual on GitHub (https://github.com/vdemichev/DiaNN) for more information.
Technical Validation
Reassembly of axial elongation during myopia progression
Mice were subjected to unilateral −30 diopter spectacles treatment, while the contralateral eye served as an age-matched control group from P21. This treatment, known as lens-induced myopia (LIM), was employed to experimentally induce axial elongation in the eyes of animal models. It leads to physiological changes that result in a mismatch between refractive power and the length of the eyeball. Ocular biometric measurements were conducted on P21 (n = 11), P28 (n = 6), and P35 (n = 5) to assess the changes that occurred in the eyes. As expected, there were no significant differences in axial length between the treated and control eyes on P21. (Fig. 1a) After one week of LIM treatment, a statistically significant increase in axial length was observed at P28, with a mean difference of 31 μm (p < 0.01). (Fig. 1b) This difference further increased after 2 weeks of LIM treatment at P35, with a mean difference of 41.4 μm (p < 0.05). (Fig. 1c) Similarly, there were no significant differences in refractive error between eyes at P21. (Fig. 1d) However, a significant difference in refractive error was observed after one week of LIM treatment at P28, with a mean difference of −7.1D (p < 0.01). (Fig. 1e) This difference persisted and further developed after two weeks of LIM treatment at P35, with a mean difference of −10.1D (p < 0.01). (Fig. 1f) Statistical analysis was performed using paired Student’s t-test with Prism software (ver. 9.5). These results reassembly that unilateral LIM treatment induces significant axial elongation after one and two weeks of manipulated optical defocus, supporting the expected model of myopia and therefore the acquired library of normal and myopic retinal proteome.

Ocular biometric measurements. Scatter plots of axial length at postnatal day (a) P21, (b) P28, and (c) P35 are shown. Refractive error measurements in diopter at (d) P21, (e) P28, and (f) P35 of eyes treated with LIM (blue) and contralateral control eye (red) are presented as mean ± SEM. Statistical analyses were performed using paired Student’s t test. The statistical significance levels are indicated as * for p ≤ 0.05, ** for p ≤ 0.05, and ns for not significant.
Intra-batch performance, precision, and reproducibility
The analysis of mice retinas revealed 6,586 unique protein groups quantified with a 1% false discovery rate (FDR), accounting for 38% of the reference proteome (UP000000539) containing 17,173 reviewed proteins annotated in the UniProt database. (Fig. 2a) There were 68,396 unique peptide sequences, with 93,688 precursors identified in the mice retina. (Fig. 2b) The in-depth analysis allowed for consistent quantification of the retinal proteome, with an average of 6,263 ± 86 unique protein groups in individual mice retinas. (Fig. 2c) These results suggest that the method preserved the quantitative features in the majority of proteins. The software computed 1% FDR cut-off was applied to individual samples, resulting in the identification of 5,512 out of 6,586 (83.7%) proteins in all samples acquired in duplicate. (Fig. 2d) The coefficient of variation (CV) analysis demonstrated that proteins quantified with more or equal to 3 peptides had the least variation with a mean value of 6.6% and median of 5.1%. Proteins with only 2 peptides showed an increased %CV with a mean value of 16.5% and median of 15.8%, while proteins quantified with only 1 peptide had a mean %CV of 18% and median of 16.5%. (Fig. 2e) To evaluate quantitative robustness, variance analysis was performed, and it was found that 6,190 (94%) of the proteins quantified had CV values below the 20% threshold. (Fig. 2f) The distribution of protein fold-change between technical replicates varied depending on the number of identified peptides. Proteins quantified with ≥3 peptides exhibited the least variation (Log2FC ± SD: −0.02 ± 0.07), while proteins quantified with 2 peptides showed slightly higher variation (Log2FC ± SD: −0.04 ± 0.16) and proteins quantified with only 1 peptide showed the largest variation (Log2FC ± SD: −0.02 ± 0.22). (Fig. 2g,h) The method quantified 5,584 (84.8%) proteins with ≥3 peptides, 609 (9.2%) proteins with 2 peptides and 393 (6%) proteins with only 1 peptide. (Fig. 2i) These results highlight the high proteome coverage and robust quantification achieved through the library-free approach for proteins with ≥3 peptide identities per protein. Among the 93,688 precursors, a range of charges from +1 to +4 was observed, with a predominance of +2 and +3 charges. (Fig. 2j) The peptide length ranged from 7 to 26 amino acids, with peptide masses between 612 and 2822 Da, and precursor m/z values between 315 and 1101. (Fig. 2k–m) Finally, the protein fold-change histogram revealed that most proteins (5342, 81.5%) exhibited minimal differences between technical replicates, with fold-change values (Log2) ranging from −0.1 to 0.1. (Fig. 2n) The uniformly distributed protein abundances in retinal tissues acquired in technical duplicate demonstrated that there were no significant differences in intra-batch variation between acquisitions. (Fig. 3a) Similarly, there was no significant distortion of the average protein intensity in every retinal tissue. (Fig. 3b) Finally, the protein fold-change demonstrated that the majority of proteins were unchanged with Log2FC value ranging from −0.104 to +0.101 in the 5–95% percentile range. (Fig. 3c) These findings provide valuable insights into the retinal proteome associated with lens-induced myopia, demonstrating robustness and coverage of the quantitative proteomics approach.

Characteristics of retinal proteome quantification. (a) Total quantified unique protein groups in the reference proteome, (b) Total quantified unique peptides in precursors, (c) Number of protein groups quantified in each sample at 1% FDR, (d) Number of protein groups according to sample coverage with missing values, (e) Variance analysis of all quantified proteins and groups of protein quantified with ≥3, 2, and only 1 peptide, (f) Number of proteins at specific %CV thresholds, (g) Normal distribution of each protein fold-change in technical duplicates, (h) Protein fold-change represented as mean ± SD by peptide groups, (i) Number of identified peptides per protein, (j) Precursor charges, (k) Peptide length, (l) Peptide monoisotopic mass, (m) Peptide m/z values, (n) Histogram of protein fold-change.

Distribution of protein abundances and fold-change. Box plots of (a) Technical replications of each acquired samples, (b) The averaged protein intensity of each retinal tissues from technical replications, (c) Protein fold-change of the paired LIM-treated eye and contralateral control eye in an individual C57BL/6 J mice.
Technical validation of the retinal-specific peptide-centric spectral library
The generation of a comprehensive spectral library specific to mice retinas using DIA-NN enables accurate identification and precise quantification of proteins in independent experiments. The library consists of a total identification of 9,401 protein groups, 70,041 peptides, 95,339 precursors, and 761,868 transitions. (Fig. 4a,b) The quantified proteins are distributed throughout the retinal proteome expression level, as demonstrated by ranking the protein abundance. (Fig. 4c) The histogram shows the distribution of the variation of individual proteins, with a median value of 6%. (Fig. 4d) Moreover, the method maintains sequence coverage of protein with a median value of 5 peptides per protein identification. (Fig. 4e) Finally, the peptide-centric method was capable of quantifying 5512 proteins in an individual sample, with only a fraction of proteins missing values in all samples. (Fig. 4f) This highlights the peptide-centric workflow using DIA-NN in capturing a more comprehensive view of the retinal proteome.

Rank and distribution of quantified proteins in spectral library. (a) Number of protein in spectral library, (b) Number of quantified proteins in spectral library, (c) Protein abundance, (d) Distribution of the coefficient of variation of protein, (e) Distribution of the number of quantified peptides per protein identification, (f) Distribution of proteins by percentage sample coverage. Dash line (red) indicates the median value of the dataset.
Code availability
No custom computer codes were generated in this work.
References
Williams, K. M. et al. Increasing Prevalence of Myopia in Europe and the Impact of Education. Ophthalmology 122, 1489–1497, https://doi.org/10.1016/j.ophtha.2015.03.018 (2015).
Morgan, I. G. et al. IMI Risk Factors for Myopia. Invest Ophthalmol Vis Sci 62, 3, https://doi.org/10.1167/iovs.62.5.3 (2021).
Choi, J. et al. Spatial organization of the mouse retina at single cell resolution by MERFISH. Nat Commun 14, 4929, https://doi.org/10.1038/s41467-023-40674-3 (2023).
Zhang, D. et al. All-trans retinoic acid stimulates the secretion of TGF-beta2 via the phospholipase C but not the adenylyl cyclase signaling pathway in retinal pigment epithelium cells. BMC Ophthalmol 19, 23, https://doi.org/10.1186/s12886-018-1017-6 (2019).
Li, Y. et al. PI3K/AKT/mTOR signaling participates in insulin-mediated regulation of pathological myopia-related factors in retinal pigment epithelial cells. BMC Ophthalmol 21, 218, https://doi.org/10.1186/s12886-021-01946-y (2021).
Li, J. et al. Hypoxia induces beta-amyloid in association with death of RGC-5 cells in culture. Biochem Biophys Res Commun 410, 40–44, https://doi.org/10.1016/j.bbrc.2011.05.101 (2011).
Wang, Q. et al. The Effect of Low-Dose Atropine on Alpha Ganglion Cell Signaling in the Mouse Retina. Front Cell Neurosci 15, 664491, https://doi.org/10.3389/fncel.2021.664491 (2021).
Liu, Y., Wang, X., Gong, R., Xu, G. & Zhu, M. Overexpression of Rhodopsin or Its Mutants Leads to Energy Metabolism Dysfunction in 661w Cells. Invest Ophthalmol Vis Sci 63, 2, https://doi.org/10.1167/iovs.63.13.2 (2022).
Bian, J. et al. SWATH Based Quantitative Proteomics Reveals Significant Lipid Metabolism in Early Myopic Guinea Pig Retina. Int J Mol Sci 22, https://doi.org/10.3390/ijms22094721 (2021).
Senabouth, A. et al. Transcriptomic and proteomic retinal pigment epithelium signatures of age-related macular degeneration. Nat Commun 13, 4233, https://doi.org/10.1038/s41467-022-31707-4 (2022).
Pan, L. et al. Baicalein-A Potent Pro-Homeostatic Regulator of Microglia in Retinal Ischemic Injury. Front Immunol 13, 837497, https://doi.org/10.3389/fimmu.2022.837497 (2022).
Kwong, J. M. K. et al. Differential Retinal Protein Expression in Primary and Secondary Retinal Ganglion Cell Degeneration Identified by Integrated SWATH and Target-Based Proteomics. Int J Mol Sci 22, https://doi.org/10.3390/ijms22168592 (2021).
Cheung, C. Y. et al. A deep learning model for detection of Alzheimer’s disease based on retinal photographs: a retrospective, multicentre case-control study. Lancet Digit Health 4, e806–e815, https://doi.org/10.1016/S2589-7500(22)00169-8 (2022).
Lopez-Cuenca, I. et al. The relationship between retinal layers and brain areas in asymptomatic first-degree relatives of sporadic forms of Alzheimer’s disease: an exploratory analysis. Alzheimers Res Ther 14, 79, https://doi.org/10.1186/s13195-022-01008-5 (2022).
Alber, J. et al. Retina pathology as a target for biomarkers for Alzheimer’s disease: Current status, ophthalmopathological background, challenges, and future directions. Alzheimers Dement https://doi.org/10.1002/alz.13529 (2023).
Javitt, D. C. et al. Disruption of early visual processing in amyloid-positive healthy individuals and mild cognitive impairment. Alzheimers Res Ther 15, 42, https://doi.org/10.1186/s13195-023-01189-7 (2023).
Remy, I. et al. Association between retinal and cortical visual electrophysiological impairments in schizophrenia. J Psychiatry Neurosci 48, E171–E178, https://doi.org/10.1503/jpn.220224 (2023).
Wagner, S. K. et al. Association Between Retinal Features From Multimodal Imaging and Schizophrenia. JAMA Psychiatry 80, 478–487, https://doi.org/10.1001/jamapsychiatry.2023.0171 (2023).
Chen, J., Chen, D. F. & Cho, K. S. The Role of Gut Microbiota in Glaucoma Progression and Other Retinal Diseases. Am J Pathol 193, 1662–1668, https://doi.org/10.1016/j.ajpath.2023.06.015 (2023).
Wolf, J. et al. Liquid-biopsy proteomics combined with AI identifies cellular drivers of eye aging and disease in vivo. Cell 186, 4868–4884 e4812, https://doi.org/10.1016/j.cell.2023.09.012 (2023).
Ludwig, C. et al. Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol Syst Biol 14, e8126, https://doi.org/10.15252/msb.20178126 (2018).
Collins, B. C. et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat Commun 8, 291, https://doi.org/10.1038/s41467-017-00249-5 (2017).
Batth, T. S., Francavilla, C. & Olsen, J. V. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J Proteome Res 13, 6176–6186, https://doi.org/10.1021/pr500893m (2014).
Chan, K. C. & Issaq, H. J. Fractionation of peptides by strong cation-exchange liquid chromatography. Methods Mol Biol 1002, 311–315, https://doi.org/10.1007/978-1-62703-360-2_23 (2013).
Searle, B. C. et al. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat Commun 9, 5128, https://doi.org/10.1038/s41467-018-07454-w (2018).
Sze, Y. H. et al. High-pH reversed-phase fractionated neural retina proteome of normal growing C57BL/6 mouse. Sci Data 8, 27, https://doi.org/10.1038/s41597-021-00813-1 (2021).
Van Puyvelde, B. et al. Removing the Hidden Data Dependency of DIA with Predicted Spectral Libraries. Proteomics 20, e1900306, https://doi.org/10.1002/pmic.201900306 (2020).
Searle, B. C. et al. Generating high quality libraries for DIA MS with empirically corrected peptide predictions. Nat Commun 11, 1548, https://doi.org/10.1038/s41467-020-15346-1 (2020).
Yang, Y. et al. In silico spectral libraries by deep learning facilitate data-independent acquisition proteomics. Nat Commun 11, 146, https://doi.org/10.1038/s41467-019-13866-z (2020).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat Methods 17, 41–44, https://doi.org/10.1038/s41592-019-0638-x (2020).
Zhang, F. et al. A Comparative Analysis of Data Analysis Tools for Data-Independent Acquisition Mass Spectrometry. Mol Cell Proteomics 22, 100623, https://doi.org/10.1016/j.mcpro.2023.100623 (2023).
Zhong, C. Q. et al. Generation of a murine SWATH-MS spectral library to quantify more than 11,000 proteins. Sci Data 7, 104, https://doi.org/10.1038/s41597-020-0449-z (2020).
Palmowski, P. et al. The Generation of a Comprehensive Spectral Library for the Analysis of the Guinea Pig Proteome by SWATH-MS. Proteomics 19, e1900156, https://doi.org/10.1002/pmic.201900156 (2019).
Qin, G., Zhang, P., Sun, M., Fu, W. & Cai, C. Comprehensive spectral libraries for various rabbit eye tissue proteomes. Sci Data 9, 111, https://doi.org/10.1038/s41597-022-01241-5 (2022).
HaileMariam, M. et al. S-Trap, an Ultrafast Sample-Preparation Approach for Shotgun Proteomics. J Proteome Res 17, 2917–2924, https://doi.org/10.1021/acs.jproteome.8b00505 (2018).
Cox, J. et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13, 2513–2526, https://doi.org/10.1074/mcp.M113.031591 (2014).
Jiang, X. et al. A highly efficient murine model of experimental myopia. Sci Rep 8, 2026, https://doi.org/10.1038/s41598-018-20272-w (2018).
Schaeffel, F., Burkhardt, E., Howland, H. C. & Williams, R. W. Measurement of refractive state and deprivation myopia in two strains of mice. Optom Vis Sci 81, 99–110, https://doi.org/10.1097/00006324-200402000-00008 (2004).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 50, D543–D552, https://doi.org/10.1093/nar/gkab1038 (2022).
Sze, Y. H. et al. Deep Quantitative Spectral Library of Normal and Myopic Mice Retinas: Proteomics Data from SWATH-MS and DIA-NN. PRIDE https://identifiers.org/pride.project:PXD046983 (2024).
Acknowledgements
This study was partly funded by the InnoHK initiative, the Hong Kong Special Administrative Region Government; the Research Centre for SHARP Vision (P0043871; P0039545); General Research Fund (GRF) (15104819; 151060/18 M); Research Impact Fund (R5032-18); Shenzhen Science and Technology Innovation Commission (JCY20180507183409601); The Hong Kong Polytechnic University and the RGC-CRF equipment grant (C5033-19E).
Author information
Authors and Affiliations
Contributions
Y.H.S. and T.C.L. conceptualized the experiment. Y.H.S. performed the mass spectrometry experiments and analysis. Y.H.S., Z.B. performed the animal experiments. D.Y.Y.T., K.K.L., Q.Z., X.Y., K.T., T.K. and T.C.L provided materials. All authors reviewed the results and contributed to the paper.
Corresponding author
Ethics declarations
Competing interests
Outside the submitted work, Kazuo Tsubota reports his position as CEO of Tsubota Laboratory, Inc., a company developing myopia-related treatments. Patents have been granted for the animal myopia model (JP 6637217 in Japan and CN 110392579 B in China) and applied for internationally (WO 2018/164113 A1).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sze, Y.H., Tse, D.Y.Y., Zuo, B. et al. Deep Spectral Library of Mice Retina for Myopia Research: Proteomics Dataset generated by SWATH and DIA-NN. Sci Data 11, 1115 (2024). https://doi.org/10.1038/s41597-024-03958-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-03958-x
