
Abstract
Encarsia sophia, a heteronomous hyperparasitoid wasp, is a well-known biological control agent, but its genomic information is limited, hindering molecular investigations and understanding of multitrophic interactions. In this study, we present a chromosome-level genome assembly for E. sophia using Illumina, PacBio HiFi, and Hi-C technologies. The assembled genome size is 398.3 Mb, with a contig N50 of 1.0 Mb and a scaffold N50 of 74.0 Mb. The BUSCO completeness score is 97.1%, and genome coverage reaches 99.1%. Utilizing Hi-C assisted assembly, the genome was organized into five chromosomes, with a mounting rate of 95.1%. Repetitive sequences make up 54.6% of the genome, and 14,914 protein-coding genes were predicted, with 95.5% functionally annotated. The high-quality genome assembly of E. sophia is a significant achievement, marking the first complete genome for a heteronomous hyperparasitoid wasp. This milestone offers valuable insights into the evolution and host interactions of heteronomous hyperparasitoids, laying the foundation for extensive research in biological control.
Similar content being viewed by others

Background & Summary
The Hymenoptera, one of the four largest orders in the class Insecta, is one of the most species-rich groups of insects. With the advancement of sequencing technologies, this order has become a hotspot in insect genomics research1,2. Currently, the number of sequenced Hymenoptera genomes has reached 557 (on April 2024, based on statistics from NCBI), with 388 species sequenced in the past three years, and annotation information submitted for 125 species. Among these sequenced Hymenoptera species, 258 belong to parasitoids, primarily including 36 species of Cynipoidea, 75 species of Chalcidoidea, 98 species of Ichneumonoidea, 42 species of Proctotrupoidea, 6 species of Chrysidoidea, and 1 species of Orussidea.
Encarsia sophia (Hymenoptera: Aphelinidae) is a dominant parasitoid of the “super pest” Bemisia tabaci (Hemiptera: Aleyrodidae), serving as a crucial biological control agent against global populations of whiteflies due to its remarkable parasitic and destructive capabilities on the host3,4,5. The reproductive strategy of this parasitoid is rather unique, being a typical heteronomous hyperparasitoid. Males and females develop heteronomously, obtaining their nutritional resources from different host insects. Females, the primary parasitoids, arise from fertilized eggs and parasitize directly within the target host insect, feeding on the larvae or nymphs of the host to complete their development. Conversely, males, arising from unfertilized eggs, act as hyperparasitoids and can only parasitize secondary hosts, i.e., those already parasitized by the primary parasitoids, feeding on the larvae of the primary parasitoids to complete their development6,7,8,9. Here, mated female E. sophia parasitize directly within the nymphs of the B. tabaci, laying fertilized eggs that develop into female offspring, serving as primary parasitoids. Unmated females, on the other hand, can only parasitize secondary hosts, laying unfertilized eggs within the nymphs of conspecific or heterospecific parasitoids already parasitized within the whitefly nymphs, producing male offspring, acting as hyperparasitoids10,11. So far, no genome of a heteronomous parasitoid has been reported. In order to gain deeper insights into the characteristics of such parasitoids, we conducted whole-genome sequencing and chromosomal-level assembly of E. sophia using Illumina, PacBio, and Hi-C technologies.
Methods
Parasitoid Wasp Collection and Sequencing
Encarsia sophia population, introduced in 2008 from the Vegetable Pest Integrated Management Laboratory at Texas A&M University, USA. They were reared in the insectarium of the Laboratory of Biological Invasion Research at the Langfang Research and Development Base of the Chinese Academy of Agricultural Sciences, using cotton plant B. tabaci nymphs as hosts (26 ± 1 °C, RH65 ± 5%, light cycle 14 L:10D). The B. tabaci laboratory population originates from the MEAM1 population maintained by the Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences (CAAS), in a greenhouse at the Institute of Plant Protection, CAAS, with no history of pesticide use. The cotton variety used is CCRI 49. E sophia is a typical heteronomous hyperparasitoid with a unique reproductive strategy: females act as primary parasitoids, parasitizing first- to fourth-instar B. tabaci nymphs (primary hosts). In contrast, solitary females produce male offspring, acting as secondary parasitoids parasitizing conspecific or heterospecific parasitoid larvae inside B. tabaci nymphs (secondary hosts). Given that males are secondary parasitoids, we collected newly emerged females for sequencing. To obtain newly emerged parasitoids, we used insect pins to transfer females from black pupae to centrifuge tubes (1.5 mL). We conducted daily checks for newly emerged adults and collected a total of 4,000 females for DNA extraction using the QIAamp DNA Mini Kit (QIAGEN). Following extraction, the purity, concentration, and integrity of the DNA were evaluated with the NanoDrop 2000&8000, Qubit Fluorometer, and Agilent 4200 Bioanalyzer, respectively.
Genome size estimation and assembly
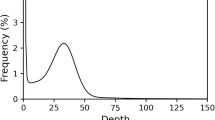
The high-quality DNA samples from E. sophia were randomly sheared using a Covaris ultrasonic disruptor. Subsequent steps, such as end repair, A-tailing, adapter ligation, purification, and PCR amplification, were performed to complete the library construction process. The constructed library was subjected to paired-end sequencing using Illumina HiSeq. By removing reads with adapter sequences and those containing more than 10% uncertain bases (N), as well as discarding single-end reads where the proportion of low-quality bases (quality score below 5) exceeds 20%, we obtained the filtered clean reads. Then, a k-mer frequency histogram was generated using Jellyfish 2.2.7 with the following parameters: “-G 2 -m 17 -C -o kmercount.”12, yielding the following estimations: a genome size of 412.21 Mbp, corrected to 404.2 Mbp, heterozygosity rate of 0.52%, and a repeat sequence proportion of 52.84% (Fig. 1). To obtain the preliminary genome assembly of E. sophia, we utilized 49,702,845,900 bp of second-generation sequencing data and assembled it using the Soapdenovo software. The assembly was then scaffolded using kmer41. The initial assembly results showed that the genome of E. sophia had a contig N50 of 1,272 bp with a total length of 318,591,742 bp, and a scaffold N50 of 2,192 bp with a total length of 328,391,604 bp (Table 1).

Encarsia sophia genome feature statistics obtained by Kmer analysis.
Sequencing was conducted using the PacBio platform, resulting in a total sequencing volume of 148 G with a coverage depth of 366.16X (calculated based on the survey-estimated genome size of 404.20 M). Additionally, a short-insert library was prepared and sequenced using the Illumina platform (Table 2). Using the sequencing data, de novo assembly of the E. sophia genome was performed with HiFiasm13. The genome assembled by Hifiasm has a length of 398.19 Mbp, with a contig N50 of 1.33 Mbp (sequences above 100 bp were selected for the assembly results) (Table 3).
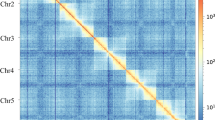
To obtain the chromosome-level genome of E. sophia, a Hi-C sequencing library was constructed using Hi-C technology14, incorporating DNA from 20,000 female adults. Hi-C data were obtained from the sequencing, and the contigs/scaffolds assembled were anchored to approximate chromosome-level using the All-hic software15. Subsequently, the juicebox software (https://github.com/aidenlab/Juicebox) was utilized for manual correction based on chromosomal interaction intensity, resulting in the final chromosome-level genome of E. sophia (Table 4). Following Hi-C-assisted assembly, the E. sophia genome assembled at the chromosome-level comprises a total of 5 sequences, with an additional 189 sequences remaining unassembled at the chromosome-level. The total genome length is 398,274,414 bp, of which 378,887,893 bp is assembled onto chromosomes (Fig. 2). The genome mapping rate achieved is 95.1% (Tables 5, 6). (Results were based on contigs above 100 bp for assembly statistics).

A genome-wide Hi-C interaction map of Encarsia sophia (5 chromosomes, 100 kb resolution) is shown, with a color gradient on the right indicating the interaction strength. Intrachromosomal interactions (red squares along the diagonal) are markedly more intense than interchromosomal interactions (light yellow squares).
Genome quality assessment
We employed different methods to assess the sequence integrity, consistency, and accuracy of the genome assembly. Firstly, the integrity of the E. sophia genome assembly was assessed using BUSCO with the insecta-odb10 database16, employing software such as MetaEuk and HMMER. The assembly resulted in 97.1% complete BUSCO genes, with 92.1% being single-copy genes and 5.0% being completely duplicated genes. Additionally, a core gene library comprising 248 conservative genes present in six eukaryotic model organisms was used for CEGMA assessment17 using tblastn, genewise, and geneid software. The assembly successfully identified 233 out of 248 core eukaryotic genes, indicating a completeness rate of 93.9%. Secondly, the sequence consistency of the E. sophia genome was assessed by aligning short-insert library reads using BWA software (http://bio-bwa.sourceforge.net/)18. The analysis revealed a HiFi reads alignment rate of approximately 97.6% and a genome coverage rate of around 99.1%, demonstrating strong consistency between the reads and the assembled genome. SNP calling was performed using samtools (http://samtools.sourceforge.net/) on the BWA alignment results, and after filtering and statistical analysis19, the genome exhibited a heterozygous SNP rate of 0.317095% and a homozygous SNP rate of 0.000943%, demonstrating excellent single-base accuracy in the assembly. Thirdly, the sequence accuracy of the E. sophia genome was assessed using Merqury software (https://github.com/marbl/merqury) with Illumina sequencing data. The quality value (Qv) of the genome, calculated based on K-mer using the Merqury-mash module20,21, was determined to be 33.6653, indicating a base accuracy rate exceeding 99.9%. In conclusion, the E. sophia genome assembly exhibits good consistency, completeness, and accuracy (Table 7).
Genome annotation
Our approach to repetitive annotation utilizes a thorough strategy that combines homology alignment with de novo search to detect repetitive sequences across the entire genome. We utilized TRF (http://tandem.bu.edu/trf/trf.html)22 for ab initio prediction, extracting tandem repeat sequences. For homology-based prediction, we utilized the standard Repbase database (http://www.girinst.org/repbase)23, employing RepeatMasker (http://www.repeatmasker.org/)24 and its internal script, RepeatProteinMask, to identify repetitive regions with default settings. In the de novo prediction process, we applied LTR_FINDER (http://tlife.fudan.edu.cn/ltr_finder/)25, RepeatScout (http://www.repeatmasker.org/), and RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html)26 to create a de novo repetitive element database. Subsequently, all repetitive sequences longer than 100 bp with an ‘N’ content below 5% were included in the initial transposable element (TE) library. This custom library, created by merging Repbase with our de novo TE library and refined using uclust to remove redundancy, was then utilized by RepeatMasker for the identification of repetitive sequences at the DNA level. The E. sophia genome contains 214.7 Mb of repetitive sequences, constituting 53.92% of the genome. Among them, long terminal repeats (LTRs) are the most abundant, accounting for 34.59% of the total, followed by Unknown (12.17%), 7.18% DNA elements, 3.96% long interspersed nuclear elements (LINEs), and short interspersed nuclear elements (SINEs) at a mere 0.02% (Table 8).
The protein-coding gene annotation in the E. sophia genome integrates de novo prediction, homology-based approaches, and RNA-Seq-supported modeling for gene prediction27. For de novo gene prediction, our automated gene prediction pipeline utilized Augustus (v3.2.3) (http://bioinf.uni-greifswald.de/augustus/)28, Geneid (v1.4), Genescan (v1.0), GlimmerHMM (v3.04) (http://ccb.jhu.edu/software/glimmerhmm/)29, and SNAP (http://homepage.mac.com/iankorf/)30. Homologous protein sequences were downloaded from NCBI Nasonia vitripennis (Nvit), Ceratosolen solmsi (Csol), Copidosoma floridanum (Cflo), Trichogramma brassicae (Tbra), Trichomalopsis sarcophagae (Tsar), Trichogramma pretiosum (Tpre). Using TblastN (v2.2.26; E-value ≤ 1e−5), protein sequences were aligned to the E. sophia genome31, and GeneWise (v2.4.1)32 software was employed to align matching proteins with homologous genomic sequences for accurate splice alignment and prediction of gene structures within each protein region. We constructed seven RNA-seq libraries, including different developmental stages of female E. sophia (600 eggs, Bemisia tabaci nymphs parasitized for <24 hours, dissected for host sampling; 200 first-instar larvae, B. tabaci nymphs parasitized for 48–60 hours, dissected for host sampling; 200 second-instar larvae, B. tabaci nymphs parasitized for 72–84 hours, dissected for host sampling; 80 third-instar larvae, B. tabaci nymphs parasitized for 120–132 hours, dissected for host sampling; 40 prepupae, B. tabaci nymphs parasitized for 168–178 hours, sampled after removing the host shell; 30 pupae, B. tabaci nymphs parasitized for 216–228 hours, sampled after removing the host shell; 50 adults, eclosed within < 24 hours.). Total RNA extracted from the aforementioned samples were used for library preparation, and sequencing was performed on the Illumina NovaSeq6000 platform33. The sequencing output generated a total of 60.51 G raw data, and after filtering, 59.88 G clean data was used for genome annotation. For genome annotation, the transcriptome was assembled using Trinity (v2.1.1)34. To refine the annotation, RNA-Seq data were processed with Hisat (v2.0.4)35 under default settings to identify exonic regions and splice sites. The alignment results were subsequently used as input for Stringtie (v1.3.3)36 with its default parameters, facilitating genome-guided transcriptome assembly. A comprehensive, non-redundant reference gene set was then created by merging the predictions from all three methods using EvidenceModeler (EVM, v1.1.1)37, which incorporated masked transposable elements for accurate gene prediction. A total of 14,914 protein-coding genes were predicted in E. sophia genome. The average length of predicted genes was 11,273.01 base pairs, with an average protein-coding region length of 1,451.53 bp. The average lengths of exons and introns were 275.58 and 2,301.66 bp, respectively. On average, each gene contained 5.27 exons (Table 9, Fig. 3).

Circular plot illustrating the chromosome-level genome assembly results for Encarsia sophia. A: chromosome information, B: gene density, C: GC content, D: ncRNA density, E: repeat density.
Gene functions were determined by aligning the E. sophia protein sequences with the Swiss-Prot database using Blastp, applying a threshold E-value of ≤1e−5 to identify the best matches. InterProScan70 (v5.31)38 was used to annotate protein motifs and domains through searches across various public databases such as ProDom, PRINTS, Pfam, PANTHER, PROSITE and SMART. Gene Ontology (GO) IDs were subsequently assigned based on the relevant InterPro entries. We mapped the genes to the NR20 database using the closest BLAST hits from the Swissprot20 database39 (E-value < 10–5) and DIAMOND (v0.8.22)/BLAST hits (E-value < 10-5). Furthermore, the genome was aligned with KEGG pathways40 to determine the best match for each gene. Ultimately, 14,245 genes (95.5% of the total) in E. sophia genome were successfully annotated in at least one database41 (Table 10).
To annotate non-coding RNAs (ncRNAs) in the E. sophia genome, tRNA genes were predicted using the tRNAscan-SE tool (http://lowelab.ucsc.edu/tRNAscan-SE/)42. Given the high conservation of rRNA sequences, we used sequences from closely related species as references and applied BLAST to identify rRNAs. Other ncRNAs, such as snRNAs and miRNAs, were detected by querying the Rfam database43 with the infernal software (http://infernal.janelia.org/)44, employing default parameters. In the end, a total of 1,457 non-coding RNAs were predicted, comprising 513 micro-RNAs (miRNAs), 514 transfer RNAs (tRNAs), 328 ribosomal RNAs (rRNAs), and 102 small nuclear RNAs (snRNAs) (Table 11).
Data Records
The sequencing data for the E. sophia genome, including Illumina, PacBio, and Hi-C datasets, have been deposited in the Sequence Read Archive (SRA) at the National Center for Biotechnology Information (NCBI) under accession numbers SRR29702816, SRR29702817, SRR2970281845,46,47, and in the Genome Sequence Archive (GSA) of the National Genomics Data Center (NGDC) under accession numbers BioProject PRJNA1131600 (NCBI) and CRA01756948 (NGDC). The transcriptome data used for annotation, covering various developmental stages of female E. sophia, have been stored in the SRA of NCBI and the GSA of NGDC: Egg (SRR2970281149, CRR1218365), 1st instar larva (SRR2970281550, CRR1218361), 2nd instar larva (SRR2970281451, CRR1218362), 3rd instar larva (SRR2970281352, CRR1218363), prepupa (SRR2970281053, CRR1218366), pupa (SRR2970280954, CRR1218367), and adult (SRR2970281255, CRR1218364). This Whole Genome Shotgun project has been deposited at GenBank under the accession JBFBOU00000000056. The genome annotation files of Encarsia sophia are available in figshare under a https://doi.org/10.6084/m9.figshare.2642675241.
Technical Validation
The quality, concentration, and integrity of the DNA samples were evaluated using a NanoDrop 2000&8000, a Qubit 3.0 Fluorometer (Thermo Fisher Scientific, USA), and an Agilent 4200 Bioanalyzer (Agilent Technologies, CA, USA), respectively. RNA integrity was assessed using the RNA Nano 6000 kit on the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). High-quality DNA and RNA were selected for library preparation and sequencing. Genome assembly integrity was verified using BUSCO (Benchmarking Universal Single-Copy Orthologs: http://busco.ezlab.org/) and CEGMA (Core Eukaryotic Genes Mapping Approach: http://korflab.ucdavis.edu/datasets/cegma/). The short-read sequences from the fragment library were mapped to the assembled genome using BWA software (http://bio-bwa.sourceforge.net/), and alignment rates, genome coverage, and depth distribution were analyzed to evaluate the completeness and uniformity of the assembly. Additionally, the genome’s quality value (Qv) was determined using the Merqury-mash module (https://github.com/marbl/merqury) to assess the sequence accuracy of the assembled genome.
Code availability
Data processing followed the standard protocols and guidelines of the relevant bioinformatics software, with default parameters applied unless specified otherwise. Details on the software versions and specific parameters are provided in the Methods section.
References
Ye, X. H. et al. A chromosome-level genome assembly of the parasitoid wasp Pteromalus puparum. Mol Ecol Resour. 20, 1384–1402 (2020).
Zhong, Y. W. et al. A chromosome-level genome assembly of the parasitoid wasp Eretmocerus hayati. Sci Data. 10, 585 (2023).
Charles, O. A review of management of major arthropod pests affecting cassava production in Sub-Saharan Africa. Crop Prot. 175, 1–15 (2024).
Katono, K. et al. Effect of Bemisia tabaci SSA1 host density and cassava genotype on host feeding capacity and parasitism by two Hymenoptera parasitoid species. Biocontrol Sci Technol. 33, 19–34 (2022).
Caspary, R. et al. Cutting Dipping Application of Flupyradifurone against Cassava Whiteflies Bemisia tabaci and Impact on Its Parasitism in Cassava. Insects. 14, 796 (2023).
Walter, G. H. Divergent male ontogenies in Aphelinidae (Hymenoptera, Chalcidoidea): A simplified classification and a suggested evolutionary sequence. Biol. J. Linn. Soc. Lond. 19, 63–82 (1983).
Mills, N. J. et al. Prospective modelling in biological control: An analysis of the dynamics of heteronomous hyperparasitism in a cotton-whitefly-parasitoid system. J Appl Ecol. 33, 1379–1394 (1996).
Williams, T. Invasion and displacement of experimental populations of a conventional parasitoid by a heteronomous hyperparasitoid. Biocontrol Sci Technol. 6, 603–618 (1996).
Hunter, M. S. et al. Evolution and behavioral ecology of heteronomous aphelinid parasitoids. Annu. Rev. Entomol. 46, 251–290 (2001).
Yang, N. W. et al. Shifting preference between oviposition vs. host-feeding under changing host densities in two aphelinid parasitoids. PLoS One. 7, e41189 (2012).
Xu, H. Y. et al. Competitive interactions between parasitoids provide new insight into host suppression. PLoS One. 8, e82003 (2013).
Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764 (2011).
Cheng, H. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods. 18, 1–6 (2021).
Belaghzal, H. et al. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods 123, 56–65 (2017).
Zhang, X. et al. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants. 5, 833–845 (2019).
Manni, M. et al. BUSCO Update: Novel and streamlined workfows along with broader and deeper phylogenetic coverage for scoring of Eukaryotic, Prokaryotic, and Viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Genis, P. et al. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Langmead, B. et al. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 9, 357–359 (2012).
Li, H. A. statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 27, 2987–2993 (2011).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Rhie, A. et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Bao, W. et al. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA. 6, 11 (2015).
Tarailo-Graovac, M. et al. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 4, 10 (2009).
Xu, Z. et al. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, 265–268 (2007).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457 (2020).
Mei, Y. et al. InsectBase 2.0: a comprehensive gene resource for insects. Nucleic Acids Res. 50, D1040–D1045 (2022).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–439 (2006).
Majoros, W. H. et al. TigrScan and GlimmerHMM: two open-source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Korf, I. Gene fnding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Birney, E. et al. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Cock, P. J. A. et al. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 38, 1767–1771 (2010).
Bolger, A. M. et al. Trimmomatic: a fexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D. et al. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Jones, P. et al. InterProScan 5: genome-scale protein function classifcation. Bioinformatics 30, 1236–1240 (2014).
Bairoch, A. et al. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 (2000).
Kanehisa, M. et al. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Man, X. Genome assembly and annotation files of Encarsia sophia. figshare https://doi.org/10.6084/m9.figshare.26426752 (2024).
Chan, P. P. et al. tRNAscan-SE 2.0: improved detection and functional classifcation of transfer RNA genes. Nucleic Acids. Res. 49, 9077–9096 (2021).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids. Res. 49, D192–D200 (2021).
Nawrocki, E. P. et al. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29702816 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29702817 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29702818 (2024).
CNCB Genome Sequence Archive https://bigd.big.ac.cn/gsa/browse/CRA017569 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29702811 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29702815 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29702814 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29702813 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29702810 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29702809 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29702812 (2024).
Man, X. Encarsia sophia isolate IPP_NW_YANG_2024, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JBFBOU000000000.1 (2024).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2023YFC2605200), the Tianshan Talent Program for Outstanding Young Scientific and Technological Talents (2022TSYCCX0084) and National Natural Science Foundation of China (32072493).
Author information
Authors and Affiliations
Contributions
N.W.Y. and W.X.L. conceived and designed the study; X.M.M. and S.H.W. collected the samples for sequencing; X.M.M. and C.H. performed the data analysis; X.M.M. wrote the draft manuscript; X.M.M., C.H., J.Y.G., F.H.W., W.X.L., N.W.Y. and F.F. discussed the results and improved and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Man, X., Huang, C., Wu, S. et al. A chromosome-level genome assembly of the heteronomous hyperparasitoid wasp Encarsia sophia. Sci Data 11, 1250 (2024). https://doi.org/10.1038/s41597-024-04040-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-04040-2
