
Abstract
Lagerstroemia speciosa is a famous medicinal and ornamental plant of the Lagerstroemia genus, with large and gorgeous flowers and a flowering period of 3 to 5 months. L. speciosa extracts have been used for many years in folk medicine to treat diabetes. Here, we used PacBio and Hi-C sequencing technologies to obtain a high-quality whole genome map of L. speciosa at chromosome level. The assembled genome is 306.76 Mb, with a scaffold N50 of 13.03 Mb. 98.75% of contigs were anchored to 24 pseudochromosomes, and 38.58% of the contigs (118.11 Mb) were identified as repeats. 93.54% of the 29,351 protein-coding genes were annotated. In addition, 146 miRNAs, 511 tRNAs, 2,733 rRNAs and 679 snRNAs were annotated. This high-quality genome assembly provides a valuable resource for understanding the species evolution in the Lythraceae family and promoting the study of important traits of L. speciosa.
Similar content being viewed by others

Background & Summary
Lagerstroemia speciosa, a species within the genus Lagerstroemia and the Lythraceae family, originates from India and Oceania, and exhibits a widespread distribution across tropical and subtropical regions1. L. speciosa prefers warm and humid climates with strong resistance and is a typical summer flowering plant with long flowering periods, large flowers and bright colors2. The breeding goal of L. speciosa is to enrich its color and flower pattern, improve its ornamental characteristics, and to apply it in various landscaping applications3. L. speciosa possesses robust fertility, boasts a high survival rate for cuttings, and is capable of interspecific hybridization with numerous Lagerstroemia species4. At present, studies on L. speciosa primarily focus on the exploration of its medicinal properties, with over 40 compounds discovered in its foliage5,6. Numerous scientific investigations have demonstrated that extracts from L. speciosa exhibit remarkable benefits in facilitating bodily functions and enhancing overall health, positioning it as a promising candidate for future therapeutic research on biologically active plant components7,8.
With the recent advancements in PacBio and Hi-C sequencing technologies, we have achieved a high-quality chromosome-level assembly of L. speciosa. The whole-genome assembly size of L. speciosa is 306.76 Mb, which was anchored to 24 chromosomes, with a scaffold N50 of 13.03 Mb and an impressive 98.75% mapping rate. The successful assembly of the genome will greatly facilitate research on the ornamental characteristics of L. speciosa and molecular-assisted breeding efforts.
Methods
Samples collection and sequencing
The diploid superior individual of L. speciosa were used as sequencing materials (Fig. 1A), which were obtained from the wild germplasm collected from the nursery of Guangxi Forestry Research Institute, located in Nanning, China (22°92′ N, 108°36′ E). The fresh young leaves were collected and soaked in liquid nitrogen immediately, then stored at −80 °C for subsequent genome sequencing and Hi-C analysis.

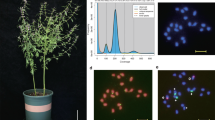
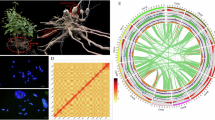
Characteristics of L. speciosa genome assembly. (A) Inflorescence of L. speciosa. (B) Genome survey results based on K-mer analysis. (C) Chromosomal interaction heatmap of L. speciosa. Chr1 to Chr24 are marked with blue boxes from top to bottom. Red bar represents interaction intensity. (D) Circos map of L. speciosa genome. The tracks from outside to inside are: 24 chromosomes length (a), gene density (b), GC content (c), distribution of unknown bases (d) and syntenic blocks (e).
Genomic DNA was isolated from these samples according to the CTAB method and its concentration and purity were evaluated by NanoDrop 2000 (Thermo Scientific, USA) and gel electrophoresis. The short-read WGS sequencing data were obtained using the Illumina HiSeq2000 platform and generated approximately 290.3 Gb of data. The long-read sequencing data were sequenced on the PacBio platform, which is based on the Single Molecule Real Time (SMRT) sequencing technology, generating a total of 18.39 Gb of sequencing data. Before genome assembly, the frequencies of 17-mer were generated by Jellyfish9 (v2.2.10) based on the clean data and used for the genome evaluation by GenomeScope10. The analysis results showed that the expected genome size is approximately 349.89 Mb, with a repeat rate of about 43.65% and a heterozygosity rate of about 1.03% (Fig. 1B). Hi-C libraries were all sequenced on the BGISEQ-500 platform at the Qingdao Huada Gene Research Institute, corresponding to approximately 62.24 Gb of sequencing data. Hi-C data can assist in the scaffolding process of genome assembly. The total RNA was extracted using OminiPlant RNA Kit. The Oligo (dT) beads facilitated the enrichment of the isolated mRNA, which was subsequently fragmented into shorter segments. The cDNA fragments were sequenced on an Illumina HiSeq4000 platform. The full-length RNA sequencing was performed on the SMRTbell DNA libraries. Three libraries of different lengths were constructed using the SMARTerTM PCR cDNA Synthesis Kit. The library sizes were verified using Qubit2.0 and Agilent 2100. During sequencing, raw polymerase reads were obtained from each Zero-Mode Waveguide single-molecule sequencing reactor. These polymerase reads were then processed to yield the final read of insert or Circular Consensus Sequence (CCS). The cDNA libraries were sequenced using the PacBio RSII sequencing platform, and the raw data underwent filtering to remove redundancy.
Genome assembly
A draft contig-level genome was initially assembled using CANU (v1.8) and PacBio sequencing data. The Pilon11 (v1.23) was used to polish the preliminary assembly with short-read data through two iterations. The genome assembly reached a contig level of 306.17 Mb, with a contig N50 of 808.25 Kb. Subsequently, HiC-Pro (v2.8.0)12 software was used to filter and process the unassembled data. Based on the interaction relationship between chromosome spatial locations, the use of endonuclease to capture interaction regions and sequencing technology. The closer the chromosome regions are, the stronger the interaction becomes. This allows the scaffold to be sequenced and oriented and assembled at the chromosomal level. After data quality control, available data were assembled using Juicer (v1.5)13 and 3D-DNA14 software, and the results were compared with the initial assembly. According to the statistical file of chromosome interaction intensity, the heat map of chromosome interaction was drawn by Juicebox (v1.11.08)15 software (Fig. 1C) based on the default processing. The alignment rate between the reads captured by Hi-C and the initially assembled genome was approximately 73.48%. The complete genome assembly of L. speciosa had a size of 306.758 Mb, including 367 scaffolds and 1,539 contigs. The N50 length was 13.03 Mb, while the maximum length was 18.28 Mb (Table 1). After Hi-C-assisted assembly, the total length of sequences on chromosomes represented 98.75% of the genome, which were assembled into 24 chromosomes ranging from 9.3 Mb to 18.28 Mb (Fig. 1D & Supplementary Table 1). In terms of embryophyta_odb10 reference gene concentration, BUSCO analysis revealed a completeness of 92.8%, with 83.2% of single-copy BUSCOs, 9.6% of multicopy BUSCOs, and 1.9% of fragmented BUSCOs (Table 2).
Genome annotation
Genome annotation primarily encompasses repeat sequence annotation, gene structure annotation, gene function annotation and ncRNA annotation. Repeat sequence annotation combines both homology-based annotation and de novo annotation methods. Homology-based annotation is performed using software such as RepeatProteinMask/RepeatMasker (v4.0.9)16, based on the RepBase17,18 database (http://www.girinst.org/repbase). Additionally, genome self-sequence alignment is used with software like RepeatModeler (v1.0.11)19, Piler20 and RepeatScount, while software TRF21 and LTR-Finder22 are employed based on the intrinsic characteristics of repetitive sequences. The results showed that approximately 118.10 Mb of the repeat sequence in L. speciosa accounted for 38.58% of the whole genome. The repetitive sequence that occupies the highest proportion is the long terminal repeats (LTRs), with a length exceeding 65.62 Mb, accounting for 21.43% of the total. Furthermore, the repetitive sequences also include 21.16 Mb (6.91%) of DNA transposons, 13.66 Mb (4.46%) of long interspersed elements (LINE) and 0.016 Mb (0.053%) of short interspersed nuclear elements (SINEs) (Table 3).
Gene structure annotation was combined with various methods. Firstly, Augustus (v3.3.4)23, Genscan24 and GlimmerHMM (v3.0.4)25 software were used for de novo prediction. Then, homology-based annotation was performed using closely related sequenced plants, including Punica granatum, Eucalyptus grandis, Arabidopsis thaliana, Brassica napus, Camelina sativa, Cucumis melo, Eutrema salsugineum, Gossypium ramondii, Raphanus sativus and Theobroma cacao. RNA-seq data were compared with StringTie (v2.1.6)26,27 and HISAT2 (v2.2.0)28 to complement and refine the predicted gene set. Finally, all these annotation results were integrated and screened by EVM (v1.1.1)29, resulting in 31,378 genes with an average CDS length of 1.4 kb (Supplementary Table 2). Using BUSCO to evaluate the completeness of gene structure annotation, 92.1% of single-copy genes were fully annotated.
Using BLASTp with an E-value cutoff of 1E-5, the proteins in the gene set were functionally annotated using databases such as SwissProt, TrEMBL30, KEGG31, InterPro32 and GO33. A total of 93.54% of the genes in the genome of L. speciosa were successfully predicted, and 93.31% of the genes were annotated in the TrEMBL annotation library (Table 4).
Based on the structural characteristics of tRNA, the tRNAscan-SE (v1.4)34 software is utilized to identify tRNA sequences within the genome. rRNA is highly conserved, and rRNA sequences of closely related species can be selected as reference sequences to search for rRNA in the genome by performing BLASTN (v2.2.26) with an E-value of 1E-5. Additionally, covariance models from the Rfam35 are employed to predict miRNA and snRNA sequence information in the genome using the Infernal (v1.0)36 software, a dedicated tool for predicting non-coding RNA (Table 5).
Data Records
The raw sequencing data and genome assembly of L. speciosa have been submitted to the National Center for Biotechnology Information (NCBI). The PacBio sequencing data were deposited in the Sequence Read Archive at NCBI under accession number SRP49438137. The illuminate raw data were accessible via accession numbers SRP49493638. Hi-C sequencing data were deposited in the Sequence Read Archive at NCBI under accession number SRP49431339. The second-generation RNA-seq data were deposited in the Sequence Read Archive under accession number SRP17640040. The full-length transcriptome data were deposited in the NCBI database under accession number SRP52850141. The genome assembly have been deposited at GenBank under accession number GCA_037672795.142. The dataset of gene annotation, CDS sequences and protein sequences have been deposited at Figshare (https://doi.org/10.6084/m9.figshare.26861248)43.
Technical Validation
To verify the integrity and accuracy of the assembled chromosomal level genome, we completed a BUSCO analysis using the embryophyta_odb10 dataset to assess the integrity of the assembly. In L. speciosa, a total of 92.8% of BUSCO was found intact, indicating a relatively complete and high-quality genome (Table 2). From the heatmap of chromosome interaction, it can be seen that the interaction intensity within the same chromosome is obviously stronger than that between chromosomes, and the chromosome boundary is more obvious, indicating that the pairing effect is better and the auxiliary assembly effect was ideal.
Code availability
No specific script was utilized during the study. Instead, all commands and pipelines related to data processing were executed strictly following the manuals and protocols provided by the respective bioinformatics software tools.
References
Sharmin, T., Rahman, M. S. & Mohammadi, H. Investigation of biological activities of the flowers of Lagerstroemia speciosa, the Jarul flower of Bangladesh. BMC Complementary and Alternative Medicine 18, 231 (2018).
Hu, L. et al. Transcriptome analysis during floral organ development provides insights into stamen petaloidy in Lagerstroemia speciosa. Plant Physiology and Biochemistry 142, 510–518 (2019).
Yang, L. C. et al. Overexpression of two MADS-Box genes from Lagerstroemia speciosa causes early flowering and affects floral organ development in Arabidopsis. Agronomy 13, 976 (2023).
Pounders, C., Sakhanokho, H. & Rinehart, T. Evaluation of interspecific hybrids between Lagerstroemia indica and L. speciosa. HortScience 42, 1317–1322 (2007).
Chan, W. C. et al. Phytochemistry and pharmacology of Lagerstroemia speciosa: a natural remedy for diabetes. Int J Herbal Med 2, 100–105 (2014).
Hou, W. et al. Triterpene acids isolated from Lagerstroemia speciosa leaves as alpha-glucosidase inhibitors. Phytotherapy Research. 23, 614–618 (2010).
Klein, G., Kim, J., Himmeldirk, K., Cao, Y. Y. & Chen, X. Z. Antidiabetes and anti-obesity activity of Lagerstroemia speciosa. Evid Based Complement Alternat Med. 4, 401–407 (2007).
Stohs, S. J., Miller, H. & Kaats, G. R. A review of the efficacy and safety of banaba (Lagerstroemia speciosa L.) and corosolic acid. Phytotherapy Research. 26, 317–324 (2012).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 33, 2202–2204 (2017).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 9, e112963 (2014).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Durand, N. C. et al. Juicer provides a One-Click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Systems. 3, 99–101 (2016).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 25, 4–10 (2009).
Bao, W., Kojima, K. K. & Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mobile. DNA. 6, 1–6 (2015).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogentic and Genome Research. 110, 462–467 (2005).
Flynn, J. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA. 117, 9451–9457 (2020).
Edgar, R. C. & Myers, E. W. Piler: identification and classification of genomic repeats. Bioinformatics. 21, i152–158 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, 265–268 (2007).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312 (2004).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 268, 78–94 (1997).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source abinitio eukaryotic gene-finders. Bioinformatics. 20, 2878–2879 (2004).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 33, 290–295 (2015).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 12, 357–360 (2015).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using evidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 (2000).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Zdobnov, E. M. & Apweiler, R. InterProScan-an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847–848 (2001).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat Genet. 25, 25–29 (2000).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124 (2005).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29, 2933–2935 (2013).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP494381 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP494936 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP494313 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP176400 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP528501 (2024).
NCBI Genbank https://identifiers.org/ncbi/insdc.gca:GCA_037672795.1 (2024).
Wan, Z. et al. Genome assembly and annotation of banaba (Lagerstroemia speciosa L.). Figshare. https://doi.org/10.6084/m9.figshare.26861248 (2024).
Acknowledgements
This work was supported by the program for Science and Technology of Beijing (No. Z181100002418006) and Special Fund for Beijing Common Construction Project.
Author information
Authors and Affiliations
Contributions
T.C.Z., T.R.C. and Q.X.Z. conceived the study. M.C., J.W., H.T.P. and T.R.C. collected samples and coordinated sequencing. Z.T.W. analyzed the data and wrote the draft manuscript. T.C.Z. contributed substantially to the revisions. All authors made valuable contributions to the article and unanimously approved the final submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wan, Z., Zheng, T., Cai, M. et al. Chromosome-level genome assembly of banaba (Lagerstroemia speciosa L.). Sci Data 11, 1228 (2024). https://doi.org/10.1038/s41597-024-04109-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-04109-y
This article is cited by
-
A chromosomal haplotype-resolved genome assembly of Cuphea hookeriana
Scientific Data (2026)
