
Abstract
Liriomyza trifolii is an economically-significant polyphagous pest that infests plants grown in both field and greenhouse conditions. Unfortunately, the lack of genomic resources has hindered our understanding of its ecological adaptation and invasiveness. To address this, we assembled a chromosome-level genome sequence of L. trifolii using a combination of short Illumina reads, PacBio HiFi long sequencing, and Hi-C scaffolding technology. The genome size was calculated at 122.64 Mb, the scaffold N50 value was 23.84 Mb, and 96.25% of the assembled sequences mapped to five chromosomes. BUSCO analysis showed high completeness with 95.28% gene coverage. A total of 11,883 protein-coding genes were identified along with 20.60 Mb of transposable elements. In summary, the genome of L. trifolii provides a valuable genetic resource for understanding invasive pests and developing effective management strategies.
Similar content being viewed by others


Background & Summary
Liriomyza spp. (Diptera: Agromyzidae) are economically-important polyphagous insects that infest plants in both field and greenhouse conditions1. Originally from the Americas, Liriomyza has spread worldwide. The larvae create tunnels in leaves, and female adults puncture leaf tissue for oviposition. These activities decrease photosynthesis and stimulate leaf drop, which reduces crop quality and yield2,3,4 (Fig. 1).

Development cycle and damage of L. trifolii. (A–C) Different developmental stages of L. trifolii, (A) larva; (B) Pupa; (C) Adult. (D) Damage symptom of L. trifolii.
With the recent expansion of facility agriculture, the damage caused by Liriomyza spp. has become a serious problem. The three polyphagous species, L. trifolii, L. sativae, and L. huidobrensis, are invasive in China5, and recent ecological and molecular studies have shown that L. trifolii is the most competitive of the three species6,7,8. L. trifolii has continued to spread since its initial discovery in China9, but the underlying molecular mechanisms for its dominance among Liriomyza spp. remain unclear. The prevailing control strategy for managing L. trifolii is the use of insecticides10,11,12, which has led to interspecific competition, pesticide resistance and a growing need for more effective control methods8,11. Although genetic approaches for control are promising, high-quality genomic data are greatly needed to understand L. trifolii invasiveness.
This study describes the construction of a high-quality chromosome-level genome of L. trifolii by integrating PacBio high-fidelity (HiFi) and Illumina short reads with high-throughput chromosome conformation capture (Hi-C) data. The deduced genome was comprised of 166 contigs with a combined size of 122.64 Mb and a contig N50 value of 1.66 Mb. Additionally, 118.04 Mb was anchored to five chromosomes, and this resulted in a scaffold N50 value of 23.84 Mb. A total of 11,883 protein-coding genes were deduced, and 95.78% of these were annotated. Furthermore, we detected 20.12 Mb of repetitive sequences, accounting for 16.80% of the genome assembly. This high-quality genome assembly of L. trifolii described in this study provides crucial data for further research on this invasive insect pest.
Methods
Insect samples
The L. trifolii strain used in this study was derived from inbred laboratory strains and was reared on kidney beans under controlled conditions of 26 °C with a 16:8 h (light: dark) photoperiod13. To minimize sequence polymorphisms and achieve a high-quality genome assembly, samples were obtained from a single mating pair, and only newly-emerged adults were selected for sequencing.
Genome sequencing
The QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) was used to obtain genomic DNA from a single surface-sterilized, newly emerged L. trifolii adults and used for both Hi-C and PacBio HiFi sequencing. TRIzol kit was used to extract total RNA from L. trifolii and the purity and integrity of nucleic acids were measured by spectrophotometry and agarose gel electrophoresis, respectively.
The Illumina NovaSeq 6000 platform was used to generate paired-end libraries containing 350-bp fragments and sequenced as recommended by the manufacturer. Low-quality reads and adapter sequences were removed using High-Throughput Quality Control (HTQC) software (version 1.92.310)14. Genomic DNA was randomly cleaved into ~15 Kb fragments using Covaris g-TUBEs (Woburn, MA, USA) and purified with 0.45 × AMPure® PB magnetic beads (Beckman Coulter, Brea, CA, USA). DNA fractions (15–18 Kb) were recovered using the Sage ELF electrophoresis system (Sage Science, Beverly, MA). Primers were annealed to SMRTbell adapters on the DNA template, and Sequel II DNA polymerase was then allowed to bind and initiate sequencing, which was executed using 8 M SMRT cells and the Sequel II System (Biomarker Technologies Co., LTD, Beijing, China). This process yielded 5.87 Gb of circular consensus sequence (CCS) reads with mean lengths of 14.5 kb, resulting in 53 × coverage of the L. trifolii genome. Standard protocols15 were used to construct Hi-C libraries, and these were sequenced on the Illumina NovaSeq 6000 platform, resulting in 11.60 Gb of 150 bp paired-end clean reads.
Assembly of genome and survey of characteristics
A survey of genome characteristics is critical for assessing genome size and heterozygosity. Frequencies of k-mers (k = 19) were obtained and surveyed from Illumina short reads using Jellyfish v. 2.2.10 and GenomeScope v. 2.0, respectively16,17. Using this approach, the predicted size of the L. trifolii genome was 108.87 Mb, with a 30.11% repeat ratio, a 1.44% heterozygosity rate and a 31.27% GC content (Fig. S1).
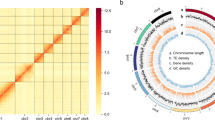
An initial assembly from PacBio long-reads of the L. trifolii genome was generated with WTDBG2 v. 2.518 using default parameters. After short reads were corrected with Pilon v. 1.2319, the L. trifolii genome was comprised of 166 contigs with a combined length of 122.64 Mb and a contig N50 of 1.66 Mb (Table S1). After removing adapter sequences and low-quality reads, 11.60 Gb of clean data were obtained and mapped to the preliminary L. trifolii genome using the Burrow-Wheeler Transform package v. 0.7.1020 with default settings. Further processing of uniquely aligned pairs was accomplished with HiC-Pro v. 2.10.021, which removes invalid read pairs, including dumped pairs, dangling ends and self-cycles. A sum of 19,398,203 valid interacting pairs were used for scaffold correction to position contigs on chromosomal DNA with LACHESIS v. 2e27abb22 and default settings. A total of 127 sequences were anchored to five chromosomes with a N50 of 23.84 Mb; this encompassed 118.04 Mb and includes 96.25% of the draft genome (Fig. 2; Table S1). Sizes of the five chromosomes ranged from 16.26–39.54 Mb (Fig. 2). Among the sequences mapped to the chromosomes, those with a determined order and orientation spanned 117.60 Mb and accounted for 99.63% of the total mapped chromosomal sequences (Table S2).

Hi-C interactive heatmap (A) and circle genome landscape (B) of L. trifolii. Color indicates the intensity of the interaction signal. The darker the color, the higher the intensity.
Annotation of repeat sequences
Repeat sequences in genomes primarily consist of tandem and interspersed repeats, with transposable elements (TEs) making up most of the latter. The repeat TE sequences in the L. trifolii genome were annotated with de novo and homology-based approaches. First, RepeatModeler v. 2.0.2a23 and LTR_retriever v. 2.824 with default settings were used to customize a de novo repeat library. The predicted repeats were then categorized with the PASTE Classifier v. 1.025 and integrated with the Dfam database v. 3.226 to generate a species-specific, non-redundant TE library. Transposable sequences were detected using homology searching using RepeatMasker v. 4.1023. Using this approach, 20.60 Mb of TE sequences were identified, which is 16.80% of the assembled genome (Table S3). Long terminal repeats (LTRs) were the most represented group of TEs and accounted for 6.92% of the genome, followed by LINEs (long interspersed nuclear elements) at 1.70%. Approximately 0.03% of the genome was populated with short interspersed nuclear elements, and transposons accounted for 8.14% of the entire genome (Table S3). Additionally, 14.90 Mb (12.15%) of tandem repeats were detected with MISA v. 2.127 and Tandem Repeat Finder28 (Table S3).
Gene prediction and functional annotation
Three strategies were implemented for prediction and assessment of protein-coding genes, including initial prediction with Augustus v. 2.429 and SNAP30, homologous species prediction using GeMoMa v. 1.3.131, and unigene prediction based on transcriptome data assembly with PASA v. 2.0.232. Homology-based gene prediction was conducted using protein sequences from four insect species including Bactrocera cucurbitae, Drosophila melanogaster, D. suzukii, and B. dorsalis, which were downloaded from InsectBase 2.033. EVidenceModeler v. 1.1.134 was then used to integrate the sequences into a unified gene set. A total of 11,883 protein-coding genes were annotated in the L. trifolii genome. For functional annotation, the predicted genes were analyzed against multiple databases including KOG (EuKaryotic Orthologous Groups), NR (Non-Redundant), TrEMBL and KEGG (Kyoto Encyclopedia of Genes and Genomes) using BLAST v. 2.2.3135 with a threshold setting of 1e−5. A total of 11,382 genes representing 95.78% of the predicted protein-encoding ORFs were annotated in one or more databases (Table S4). Additionally, 9,671 protein-encoding genes were assigned gene ontology (GO) terms and 9,236 mapped to one or more KEGG pathways (Table S4).
Data Records
The Hi-C, raw Illumina and PacBio HiFi sequencing data for the L. trifolii genome has been deposited in the NCBI Sequence Read Archive (SRA) database as accession number SRP51001036. The final chromosome assembly is available in the GenBank as accession no. JBHGZK00000000037. The genome annotation for L. trifolii has been uploaded to figshare (https://figshare.com/) with the identifier 2612243238.
Technical Validation
Validation of the genome assembly
Three independent methods were employed to evaluate the completeness and accuracy of the L. trifolii genome assembly. First, clean reads from Illumina sequencing were aligned to the genome assembly using Burrow-Wheeler Transform algorithm (BWA)20, and this analysis showed that 98.68% of the Illumina reads were correctly aligned with the genome assemblage. Next, the CEGMA database (e.g., Core Eukaryotic Genes Mapping Approach), which consists of 458 conserved eukaryotic genes, was used to assess the genome, and 100% (n = 458) of the genes were identified in the L. trifolii genome. Finally, genome assembly completeness was evaluated using BUSCO v. 2.516 with the insecta.odb10 database. and results showed that 95.28% (3130/3285) of the conserved BUSCO proteins were present in the L. trifolii genome. Among these, 70.05% were single copy, complete genes, 25.24% were complete and duplicated, 0.33% were fragmented, and 4.38% were not detected.
The quality of the chromosome assembly was further assessed by dividing the genome into 50 kb bins, and the intensity of interaction pairs was used to generate heatmaps. The Hi-C heatmap indicated greater interaction intensity along diagonals as compared to non-diagonal positions for the five distinct chromosomes (Fig. 2). These results demonstrate that the quality of the L. trifolii genome assembly is high.
Code availability
Software programs and pipelines were conducted as specified in the instruction manuals and published protocols of bioinformatic tools. Detailed information on software versions, code, and parameters can be found in the Methods section.
References
Spencer, K. A. Series Entomologica. In Göttingen ES (ed.), Agromyzidae (Diptera) of Economic Importance, 1st Edn. Vol. 9. Bath: The Hague Publishers, pp. 19-28 (1973).
Johnson, M. W., Welter, S. C., Toscano, N. C., Ting, P. & Trumble, J. T. Reduction of tomato leaflet photosynthesis rates by mining activity of Liriomyza sativae (Diptera: Agromyzidae). J. Econ. Entomol. 76, 1061–1063 (1983).
Parrella, M. P., Jones, V. P., Youngman, R. R. & Lebeck, L. M. Effect of leaf mining and leaf stippling of Liriomyza spp. on photosynthetic rates of chrysanthemum. Ann. Entomol. Soc. Am. 78, 90–93 (1985).
Reitz, S. R., Kund, G. S., Carson, W. G., Phillips, P. A. & Trumble, J. T. Economics of reducing insecticide use on celery through low-input pest management strategies. Agric. Ecosyst. Environ. 73, 185–197 (1999).
Kang, L. Ecology and Sustainable Control of Serpentine Leafminers. Science Press, Beijing, pp. 86-90 (1996).
Wang, H. H., Reitz, S. R., Xiang, J. C., Smagghe, G. & Lei, Z. R. Does temperature mediated reproductive success drive the direction of species displacement in two invasive species of leafminer fly? PLoS One 9(6), e98761 (2014).
Chang, Y. W. et al. Cloning and expression of genes encoding heat shock proteins in Liriomyza trifolii and comparison with two congener leafminer species. PLoS One 12(7), e0181355 (2017).
Chang, Y. W. et al. Comparative transcriptome analysis of three invasive leafminer flies provides insights into interspecific competition. Int. J. Biol. Macromol. 165, 1664–1674 (2020).
Wang, G., Guan, W. & Chen, D. H. Preliminary report of the Liriomyza trifolii in Zhongshan area. Plant Q. 21, 19–20 (2007).
Gao, Y. L., Reitz, S. R., Wei, Q. B., Yu, W. Y. & Lei, Z. R. Insecticide-mediated apparent displacement between two invasive species of leafminer fly. PLoS One 7, e36622 (2012).
Gao, Y. L., Reitz, S. R., Xing, Z. L., Ferguson, S. & Lei, Z. R. A decade of a leafminer invasion in China: lessons learned. Pest Manag. Sci. 73, 1775–1779 (2017).
Reitz, S. R., Gao, Y. L. & Lei, Z. R. Insecticide use and the ecology of invasive Liriomyza leafminer management. In Trdan S (ed.), Insecticides-Development of Safer and More Effective Technologies. Rijeka: InTech, pp. 235-255 (2013).
Chen, B. & Kang, L. Cold hardiness and supercooling capacity in the pea leafminer Liriomyza huidobrensis. Cryo-Letters. 23(3), 173–182 (2002).
Yang, X. et al. HTQC: a fast quality control toolkit for Illumina sequencing data. BMC Bioinformatics 14, 33–36 (2013).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159, 1665–1680 (2014).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212 (2015).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 33, 2202–2204 (2017).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat Methods. 17, 155–158 (2020).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS one. 9, e112963 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–1760 (2009).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 1–11 (2015).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol. 31, 1119–1125 (2013).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457 (2020).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant physiol. 176, 1410–1422 (2018).
Hoede, C. et al. PASTEC: an automatic transposable element classification tool. PLoS one. 9, e91929 (2014).
Wheeler, T. J. et al. Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res. 41, D70–D82 (2012).
Beier, S., Tiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics. 33, 2583–2585 (2017).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 24, 637–644 (2008).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics. 5, 59 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89 (2016).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Mei, Y. et al. InsectBase 2.0: a comprehensive gene resource for insects. Nucleic Acids Res. 50, D1040–D1045 (2022).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, 1–22 (2008).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP510010 (2024).
Chang, Y. W. & Du, Y. Z. Liriomyza trifolii isolate CY-2024, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JBHGZK000000000 (2024).
Chang, Y. W. Liriomyza trifolii genome. figshare https://doi.org/10.6084/m9.figshare.26122432.v1 (2024).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 32202275), National Key Research and Development Program of China (Grant No. 2022YFC2601100), and Jiangsu Agricultural Industry Technology System (Grant No. JATS [2023] 315).
Author information
Authors and Affiliations
Contributions
Y.C. and Y.D. conceived the project; Y.W., Y.W. and Y.C. performed the experiments; Y.C. performed the bioinformatic analyses; Y.C., Y.W., Y.W. and Y.D. evaluated the results; Y.C. and Y.D. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chang, YW., Wang, YC., Wang, YC. et al. Chromosome-level genome assembly of the invasive leafminer fly, Liriomyza trifolii (Diptera: Agromyzidae). Sci Data 11, 1326 (2024). https://doi.org/10.1038/s41597-024-04208-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-04208-w
