
Abstract
The deployment of liquid chromatography-mass spectrometry-based plasma proteomics experiments in a large cohort is sparse, leading to a lack of data available for benchmarking, method development or validation. Comprised of 6,426 plasma analyses, The Environmental Determinants of Diabetes in the Young (TEDDY) proteomics validation study constitutes one of the largest targeted proteomics experiments in the literature to date. The proteomics data from this study were generated over the course of 2.5 years from over 900 study subjects, each providing up to 29 longitudinal samples. The data also includes 916 quality control samples. The targeted mass spectrometry assay was comprised of 694 peptides mapping to 167 proteins and the panel was measured in each subject and QC sample. The targeted proteomic dataset presented here can be used as a resource for new computational methods development, such as for batch correction, as well as for benchmarking and comparing the performance of different methods/tools.
Similar content being viewed by others
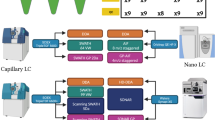
Background & Summary
Type 1 diabetes (T1D) is a complex autoimmune disease that results in the destruction of insulin-producing pancreatic beta cells and affects tens of millions of people worldwide1,2. Although there have been significant advancements in understanding the pathogenesis and treatment of T1D, there is still no cure for this disease. The Environmental Determinants of Diabetes in the Young (TEDDY) study aims to identify biomarkers of the progression of T1D, focused on the stage where i) two or more diabetes-related autoantibodies can be detected in the circulation (seroconversion) yet the subject is asymptomatic, described here as autoimmunity (IA), and ii) the subject has a clinical diagnosis of T1D. To accomplish this goal, the TEDDY study was designed to be a prospective birth cohort study of more than 8500 children genetically at risk for developing T1D, in which subjects were enrolled into the study when they are a few months old. The subjects were followed and data was collected for more than 10 years in many cases3. Multiple types of analytical measurements have been generated including but not limited to epigenomics, transcriptomics, metabolomics, and proteomics4.
An initial untargeted, liquid chromatography-mass spectrometry (LC-MS)-based proteomics study was conducted to identify candidate peptides and proteins predictive of IA and T1D by comparing control subjects to IA or T1D subjects using samples collected prior to and after seroconversion and after diagnosis of hyperglycemia, respectively. A combination of statistics and machine learning was used to identify a subset of candidate peptides of interest5. In the work presented here, a targeted plasma proteomics study of the identified candidate peptides was then conducted. Longitudinal samples from over 900 subjects were analysed, and a total of 694 peptides mapping to 167 unique proteins were selected for measurement. In addition to subject samples, hundreds of quality control (QC) samples were analysed over the course of the 2.5 years of data generation (Fig. 1). The TEDDY targeted proteomics dataset is unique in its size, the availability of a large number of QC samples, and the length of time required to analyse all samples. As such, this data offers a valuable resource for secondary data analysis. For example, the data allows for users to evaluate and characterize batch effects in a large LC-MS dataset. Further, the presence of QC samples enables the evaluation of the effectiveness of existing and newly developed batch correction algorithms. Additionally, multiple longitudinal measurements for the same individual exist in the dataset, allowing researchers to investigate inter- and intra-subject variability in targeted proteomics data. This enables a better understanding of variability for experimental design and statistical power calculation purposes.

Study sample generation. The number of samples run per day over the course of analysing all samples by month and year. The number of samples is represented by the color filling the day’s square, with days labelled by day of the month.
Methods
Study participants
All procedures were approved by the ethics committees/institutional review boards including Colorado Multiple Institutional Review Board (04-0361); Medical College of Georgia Human Assurance Committee (2004–2010)/Georgia Health Sciences University Human Assurance Committee (2011–2012)/Georgia Regents University Institutional Review Board (2013–2017)/Augusta University Institutional Review Board (2017-present) (HAC 0405380); University of Florida Health Center Institutional Review Board (IRB201600277); Washington State Institutional Review Board (2004–2012)/Western Institutional Review Board (2013–present) (20130211); Ethics Committee of the Hospital District of Southwest Finland (Dnro168/2004); Bayerischen Landesärztekammer (Bavarian Medical Association) Ethics Committee (04089); and Regional Ethics Board in Lund, Section 2 (2004–2012)/Lund University Committee for Continuing Ethical Review (2013-present) (217/2004). Consent to participate in the study were signed by the child parent or guardian. Patient information was kept by the Data Coordination Center at University of South Florida, and no identifiable information was passed to the sample analysis center or were associated with the collected data. In addition, TEDDY is monitored by an external evaluation committee formed by the National Institute of Diabetes, Digestive and Kidney Diseases (NIDDK), Bethesda, MD, U.S.A. All samples collected by TEDDY were stored in the NIDDK repository (https://repository.niddk.nih.gov/home/), and details on the organization, handling, and storage of samples is described elsewhere6. Subjects who developed IA or T1D had samples before and after seroconversion and after diagnosis of hyperglycemia, respectively, analyzed in addition to matched control samples. A fully detailed description of the study design can be found elsewhere5. Data were made available through a formal collaboration with TEDDY. Figure 2 gives the distribution of the number of time points analyzed for each patient. TEDDY study participants have higher genetic risk of developing T1D, and samples were previously matched based on clinical center, gender, and family history of T1D3. For full access to sample metadata please reference the TEDDY access page7. The availability of data is subject to approval by the TEDDY Ancillary Studies Committee.

The number of samples per patient. Most patients had multiple samples at different time points. Here the distribution of the number of time points per patient are shown for the entire cohort.
Peptide selection for liquid chromatography-selected reaction monitoring-mass spectrometry (LC-SRM-MS) assays
In the proteomics study discovery phase, a total of 376 plasma proteins were identified as differentially abundant comparing different study groups5. These 376 biomarker candidate proteins were down selected based on statistical significance at Benjamini-Hochberg adjusted p-value ≤ 0.05, resulting in 133 candidate proteins. To maximize the number of proteins to be monitored by targeted proteomics, we had additional selection criteria. The 243 significant proteins without a p-value adjustment were further searched using criteria that would select the most identifiable proteins, those with observed potential to aid in prediction using a multivariate panel, and those previously reported as associated with T1D. Additional proteins were included based on:
-
(1)
≥ 2 peptides identified per protein which each had a spectral count (SpC) ≥ 20, was present in at least 23 samples (50%), and had an unadjusted p-value < 0.005 (14 proteins; 118 of the 132 total identified proteins in this category overlapped with the 133 candidate proteins based on the adjusted p-value). In this group, we aimed to add peptides that are well detected and have higher chance to become good SRM assays,
-
(2)
≥ 2 peptides identified per protein which each had a SpC ≥ 20, detected in more than 23 samples (50% combined to machine learning analysis, carried out to determine the group of proteins that are the most predictive of each comparison, results, and having an unadjusted ≤ 0.05 (9 unique proteins; 33 of 42 total identified proteins in this category overlapped with criteria one above). In this group, we aimed to add peptides that are good predictors of specific outcomes,
-
(3)
whether they were previously described in the literature as potential T1D biomarkers (11 proteins of 32 identified from the literature that did not overlap with prior criteria). In this group, we added peptides that can be robust biomarkers because they were supported by additional studies.
This resulted in the selection of 167 proteins for validation5. To select the peptides to represent these 167 candidate proteins, a Bayesian network was developed to compute probabilities for and filter candidate peptide targets. The Bayesian network considered the optimal size (8-20 AA) and composition (lack of M), digestion (trypsin cleavage in both termini), and amino acid characteristics to predict the probability that a peptide was an appropriate target for liquid chromatography-selected reaction monitoring-mass spectrometry (LC-SRM-MS). The size and composition of a peptide, whether it was a tryptic peptide, the number of missed cleavages seen in the previous data set, the presence of impossible cleavage amino acid pairs, and the presence of difficult to cleave amino acid pairs, including lack of post-translationally modified amino acid residues. Finally, the peptides were screened for the presence of amino acid residues that are spontaneously modified or difficult to be added by chemical synthesis: M, W, Q and N.
Development of LC-SRM-MS assays
Quality control samples comprised of pooled plasma from multiple individuals from the TEDDY repository was diluted 10-fold in 8 M urea in 50 mM NH4HCO3, pH 8.0. 100 mM dithiothreitol was added to a final concentration of 10 mM and incubated for 1 h. Samples were included 37 °C with shaking at 1200 rpm as with all subsequent steps. The samples were alkylated by adding 400 mM iodoacetamide to a final concentration of 40 mM and incubating in the dark for 1 h. Samples were diluted 6-fold in 50 mM NH4HCO3, and 1 M CaCl2 to a final concentration of 1 mM and digested with 1/50 trypsin to protein mass ratio for 6 h but with shaking at 1100 rpm.
Digested samples were desalted using C18 cartridges (Strata, 25 mg/1 mL, Phenomenex). The cartridges were rinsed with 1.5 mL of methanol and equilibrated with 0.1% trifluoroacetic acid (TFA). Samples were acidified to pH 2.0 with 10% TFA and loaded to the cartridges. The cartridges were washed with 2 mL 5% acetonitrile (ACN) in 0.1% TFA, and peptides were eluted with 0.4 mL 80% ACN. Peptides were dried in a vacuum centrifuge, resuspended in milliQ water and quantified by BCA assay following the manufacturer protocol.
Standard peptides were custom synthesized by New England Peptides (then Vivitide and now Biosynth) with heavy isotopes in the C-terminal amino acid residues. MS precursor and transitions (measured fragments) for each peptide were optimized in three steps. In the first step, mixtures of 200 peptides at 1 picomole each were loaded into a 5 µL sample loop and consequently to SPE column for 2 minutes at a flow rate of 5 µL/minute with solvent C (0.1% formic acid in water)8. Peptides were separated by reversed-phase HPLC using a column packed in-house with 3-µm Jupiter C18 stationary phase (Phenomenex, Torrance, CA) into a 60-cm length of 360 µm o.d. x 75 µm i.d. fused silica capillary tubing (Polymicro Technologies Inc., Phoenix, AZ) connected to a nanoAquity UPLC® system (Waters Corporation, Milford, MA). The column was equilibrated at 300 nL/min with 99% solvent C/1% solvent D (0.1% formic acid in acetonitrile) followed by the gradient: 0:1 (time [min]:solvent D [%]), 2:8, 20:12, 75:30, 97:45, 100:95. Spectra (AGC 1 × 106) were collected from 300-1800 m/z at a resolution of 60 K followed by HCD MS/MS (resolution of 7500, AGC 5 × 104) spectra of the ten most intense ions in a LTQ Orbitrap Velos mass spectrometer (Thermo Scientific, San Jose, CA). MS/MS spectra were collected using a 2.5 Da isolation width, 45% normalized collision energy with activation time of 0.1 ms, and dynamic excluded for 45 sec (within -0.55 and 2.55 Da). Collected data were analysed with the Skyline Daily 20.1 software9, and the 6 most intense precursors/ fragments were selected. In the second step, the heavy-labelled standards were analysed in the pooled plasma on a BEH130 C18 M-class column (100 mm × 100 µm i.d.) coupled to Nano M-class UPLC (Waters)-TSQ Altis triple quadrupole mass spectrometer (Thermo Fisher Scientific). A total of 400 ng of peptides were loaded on the column with a 5-μL loop and the peptides were eluted at 400 nL/min using the following AD gradient where buffer A is 0.1% formic acid and buffer D is 0.1% formic acid in ACN. The gradient composed of (time [min]:solvent D [%]) of 0:1 2:6, 33:13, 63:22, 73:40, 78:95, 83:95. Collected SRM data were analysed with the Skyline software9. The SRM transitions were then reduced to 3 based on intensity and interference in both the heavy and light channels. In the third step, the retention time windows were optimized for the eventuality of a final analytical method were established. Heavy-labelled standard peptides were considered detected when the signal-to-noise ratio was greater than 3. The linearity of the assays was evaluated by diluting human plasma into chicken plasma.
Targeted proteomics data collection
Two digestion QC samples were used in this study. First, a QC sample was created by pooling individual TEDDY samples. Sample was created and eight aliquots were randomized across the 96-well plates. This first QC was used for post-hoc data analysis and the researchers were blinded to these QCs. To evaluate the quality of the data while they are being collected, a second QC sample comprised of commercially available pooled plasma (BioIVT) was added to each plate to enable real time QC monitoring. In total, 6,426 individual plasma samples were digested in digestion batches of approximately 80 samples plus 9 QC samples in 96-well plate format. The protein digestion procedure was performed using an EpiMotion robot (Eppendorf). Briefly, an aliquot of 5 µL of plasma was loaded into an Eppendorf 1.3 mL deep well plate which was preloaded 45 µL of 50 mM NH4HCO3 containing 8 M solution. Proteins were reduced by adding 5 µL of 100 mM dithiothreitol (Sigma) to a final concentration of 10 mM, followed by incubation at 37 °C for 1 h in a Bioshake IQ (Bulldog Bio, Inc.). Cysteine residues were alkylated by adding 5.5 µL of 400 mM iodacetamide (Sigma) and incubating in the dark at 37 °C for 1 h. Samples were then diluted 6-fold in 50 mM NH4HCO3 pH 8.0 and digested using 1:50 enzyme/substrate trypsin gold (Promega). After incubation for 6 hours at 37 °C with constant shaking, the digest was quenched by reducing the sample pH to 3.0 using 10% trifluoracetic acid (Sigma). Digested peptides were desalted by solid-phase extraction (SPE) using a Strata C18-E 25 mg/well 96-well plate (Phenomenex,) and dried in a vacuum centrifuge. After digestion the samples were spiked with standard peptides. Targeted proteomics data were collected on the TSQ Altis mass spectrometer, as described above. Data quality was assessed using the Q4SRM tool10, monitoring for the detection of standard peptides and retention time shifts10. Data were extracted with the Skyline software9; each peptide was manually inspected for proper alignment across runs, detection of both endogenous and the heavy-isotope labelled internal standard peptides, and interference of background noise.
Data Records
The raw mass spectrometry data files were deposited into and are available on MassIVE11 (ftp://massive.ucsd.edu/v05/MSV000091562/). Sample metadata information for each mass spectrometry analysis includes the sample batch, plate, run date, sample type (i.e. subject or QC), subject ID, and the subject’s age. The processed and quantified targeted proteomics data are available through the Pacific Northwest National Laboratory’s (PNNL)’s DataHub data repository (https://data.pnnl.gov/group/nodes/dataset/33861)12.
Technical Validation
Study design, batching, and randomization
Seroconversion was determined by screening for four autoantibodies against islet cells (IA-2A), insulin (IAA), glutamic acid decarboxylase (GADA) and zinc transporter 8 A (ZnT8A), as described elsewhere13. T1D was diagnosed based on blood glucose levels as described previously14.
Sample selection, batch assignment, and analysis order randomization was performed at the University of South Florida (USF), and targeted proteomics measurements were conducted at PNNL. Samples were randomized so that study endpoints and time points were properly spread throughout the batches. Figure 3 shows the number of samples analyzed for each plate, by sample type. PNNL staff were blinded to sample endpoints until the conclusion of the study.

Sample run information. The number of samples for each experimental plate for each type of sample – subject sample, pooled QC sample, and BiolVT QC sample. Points are colored by batch each of which consisted of multiple plates.
Missing values
Missing values can occur in immunoassay and MS based proteomics studies and result from several factors such as a peptide being below the limit of detection, biological factors or matrix affects, to name a few15. Untargeted proteomics studies are subject to high levels of missing values commonly ranging from 20–50%16. However, the percentage of missing data in targeted proteomics is typically much lower than in untargeted studies. In the dataset described here, 1.85% of values were missing across all 6426 analyses, including QC samples. The median percent of missing values across all peptides was 0.03%, with an interquartile range of 0.23% and a maximum percent missing for a peptide of 76.86%. A total of 139 peptides had complete observations across all samples. Figure 4 shows the percent of missing observations for each sample by analysis order (i.e., plate) and sample type. There are no systematic patterns of missingness related to analysis order or sample type.

Missing value information. The percentage of missing values, for each sample type, by plate is shown.
Quality control samples
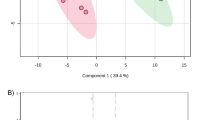
The inclusion of QC samples is a common way to monitor and assess the quality of data. Additionally, QC samples are often used to correct for variations introduced into samples over time (i.e., batch effects). Figure 5 shows the first two principal components from a sequential projection pursuit principal component analysis17 (PCA) based on peptide abundance values, which does not require imputation of missing values, on the pooled QC samples for this study. Samples from the same batch cluster together in the PCA plot indicating the presence of batch effects.

Principal component analysis of QC samples, demonstrating batch effects. The first two PCA scores for each sample based on peptide abundances, by batch, show batch effects related to time of run for the pooled quality control samples.
The coefficient of variation (CV) is a quantity (i.e., the standard deviation of measurements divided by the mean of measurements) often used to assess the quality of measurement for each peptide measured. CV values are highly dependent on the experimental design, sample media, and other factors. A more thorough discussion and evaluation of CV values in untargeted proteomics studies can be found elsewhere18. Figure 6 shows the distribution of CV values for each peptide across the pooled QC samples for this targeted proteomics study without the implementation of batch correction. The median CV value was 0.50 and the interquartile range was 0.63 indicating high reproducibility even without correcting for batch effects in the QC samples.

Coefficient of variation for QC samples. Distribution of coefficient of variation values, calculated for each peptide, for the pooled QC samples.
Usage Notes
Proteomics data should be log-transformed and normalized via methods such as global median centering. If data is desired at the protein-level, a protein quantification method such as reference-based rollup19 should be used. The R package pmartR20,21 is designed for the analysis of ‘omics data, such as proteomics, and enables all the above methodologies to be implemented on targeted proteomics datasets. If users wish to test or implement batch correction methods on the data, methods such as COMBAT22, which does not require QC samples, or Quality Control-based Robust LOESS Signal Correction (QC-RLSC)23, which does require QC samples, are among several popular methods. The R package malbacR24 has the capability to implement many batch correction methods and works with data in the same format as pmartR.
References
DiMeglio, L. A., Evans-Molina, C. & Oram, R. A. Type 1 diabetes. Lancet 391, 2449–2462, https://doi.org/10.1016/S0140-6736(18)31320-5 (2018).
Ziegler, A. G. et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 309, 2473–2479, https://doi.org/10.1001/jama.2013.6285 (2013).
Lee, H. S. et al. Biomarker discovery study design for type 1 diabetes in The Environmental Determinants of Diabetes in the Young (TEDDY) study. Diabetes Metab Res Rev 30, 424–434, https://doi.org/10.1002/dmrr.2510 (2014).
Rewers, M. et al. The Environmental Determinants of Diabetes in the Young (TEDDY) Study: 2018 Update. Curr Diab Rep 18, 136, https://doi.org/10.1007/s11892-018-1113-2 (2018).
Nakayasu, E. S. et al. Plasma protein biomarkers predict the development of persistent autoantibodies and type 1 diabetes 6 months prior to the onset of autoimmunity. Cell Rep Med 4, 101093, https://doi.org/10.1016/j.xcrm.2023.101093 (2023).
Vehik, K. et al. Methods, quality control and specimen management in an international multicentre investigation of type 1 diabetes: TEDDY. Diabetes Metab Res Rev 29, 557–567, https://doi.org/10.1002/dmrr.2427 (2013).
Group, T. S. https://teddy.epi.usf.edu/research/ (2024).
Shen, Y. et al. High-efficiency on-line solid-phase extraction coupling to 15-150-microm-i.d. column liquid chromatography for proteomic analysis. Anal Chem 75, 3596–3605 (2003).
Pino, L. K. et al. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom Rev 39, 229–244, https://doi.org/10.1002/mas.21540 (2020).
Gibbons, B. C. et al. Rapidly Assessing the Quality of Targeted Proteomics Experiments through Monitoring Stable-Isotope Labeled Standards. J Proteome Res 18, 694–699, https://doi.org/10.1021/acs.jproteome.8b00688 (2019).
Nakayasu, E. S. et al. TEDDY Proteomics Validation Phase Study. MassIVE https://doi.org/10.25345/C5KH0F84V. (2023).
Bramer, L. M. et al. TEDDY Targeted Proteomics Study Data. PNNL DataHub https://doi.org/10.25584/2371917 (2024).
Vehik, K. et al. Hierarchical Order of Distinct Autoantibody Spreading and Progression to Type 1 Diabetes in the TEDDY Study. Diabetes Care 43, 2066–2073, https://doi.org/10.2337/dc19-2547 (2020).
Vehik, K. et al. Performance of HbA1c as an early diagnostic indicator of type 1 diabetes in children and youth. Diabetes Care 35, 1821–1825, https://doi.org/10.2337/dc12-0111 (2012).
Bramer, L. M., Irvahn, J., Piehowski, P. D., Rodland, K. D. & Webb-Robertson, B. M. A Review of Imputation Strategies for Isobaric Labeling-Based Shotgun Proteomics. J Proteome Res 20, 1–13, https://doi.org/10.1021/acs.jproteome.0c00123 (2021).
Webb-Robertson, B. J. et al. Review, evaluation, and discussion of the challenges of missing value imputation for mass spectrometry-based label-free global proteomics. J Proteome Res 14, 1993–2001, https://doi.org/10.1021/pr501138h (2015).
Stacklies, W., Redestig, H., Scholz, M., Walther, D. & Selbig, J. pcaMethods–a bioconductor package providing PCA methods for incomplete data. Bioinformatics 23, 1164–1167, https://doi.org/10.1093/bioinformatics/btm069 (2007).
Piehowski, P. D. et al. Sources of technical variability in quantitative LC-MS proteomics: human brain tissue sample analysis. J Proteome Res 12, 2128–2137, https://doi.org/10.1021/pr301146m (2013).
Polpitiya, A. D. et al. DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics 24, 1556–1558, https://doi.org/10.1093/bioinformatics/btn217 (2008).
Degnan, D. J. et al. pmartR 2.0: A Quality Control, Visualization, and Statistics Pipeline for Multiple Omics Datatypes. J Proteome Res 22, 570–576, https://doi.org/10.1021/acs.jproteome.2c00610 (2023).
Stratton, K. G. et al. pmartR: Quality Control and Statistics for Mass Spectrometry-Based Biological Data. J Proteome Res 18, 1418–1425, https://doi.org/10.1021/acs.jproteome.8b00760 (2019).
Zhang, Y., Parmigiani, G. & Johnson, W. E. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom Bioinform 2, lqaa078, https://doi.org/10.1093/nargab/lqaa078 (2020).
Dunn, W. B. et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat Protoc 6, 1060–1083, https://doi.org/10.1038/nprot.2011.335 (2011).
Leach, D. T. et al. malbacR: A Package for Standardized Implementation of Batch Correction Methods for Omics Data. Anal Chem 95, 12195–12199, https://doi.org/10.1021/acs.analchem.3c01289 (2023).
Bramer, L. M., Stratton, K. G., Claborne, D., Degnan, D. J. & Webb-Robertson, B. J. pmartR Tutorials and Vignettes, https://pmartr.github.io/pmartR/index.html (2024).
Acknowledgements
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant R01 DK138335 (to ESN, BJMWR and TOM.), National Institutes of Health contract HHSN267200700014C and grant U01 DK127786 (TOM) from the National Institute of Diabetes and Digestive and Kidney Disease and U01 KD127786-S1 (BJMWR). Mass spectrometry analyses were performed in the Environmental and Molecular Sciences Laboratory, a national scientific user facility sponsored by the U.S. Department of Energy (DOE) and located on the campus of PNNL in Richland, WA. PNNL is operated by Battelle Memorial Institute for the DOE under contract DEAC05-76RLO1830. The TEDDY Study is funded by U01 DK63829, U01 DK63861, U01 DK63821, U01 DK63865, U01 DK63863, U01 DK63836, U01 DK63790, UC4 DK63829, UC4 DK63861, UC4 DK63821, UC4 DK63865, UC4 DK63863, UC4 DK63836, UC4 DK95300, UC4 DK100238, UC4 DK106955, UC4 DK112243, UC4 DK117483, U01 DK124166, U01 DK128847, and Contract No. HHSN267200700014C from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institute of Allergy and Infectious Diseases (NIAID), Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), National Institute of Environmental Health Sciences (NIEHS), Centers for Disease Control and Prevention (CDC), and Breakthrough T1D (formerly JDRF). This work is supported in part by the NIH/NCATS Clinical and Translational Science Awards to the University of Florida (UL1 TR000064) and the University of Colorado (UL1 TR002535). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
Author contributions are provided according to Contributor Roles Taxonomy (CRediT). L.M.B.: data curation, formal analysis, writing – original draft; E.S.N.: writing – original draft, writing – review & editing; J.E.F.: writing – review & editing; J.V.E.: writing – review & editing; M.J.M.: writing – review & editing; H.M.P.: writing – review & editing. T.O.M.: writing–review & editing, funding acquisition, project administration, supervision; B.J.M.W.R.: writing – review & editing, funding acquisition, project administration, supervision
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visithttp://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bramer, L.M., Nakayasu, E.S., Flores, J.E. et al. Data from a multi-year targeted proteomics study of a longitudinal birth cohort of type 1 diabetes. Sci Data 12, 112 (2025). https://doi.org/10.1038/s41597-024-04249-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-04249-1
