
Abstract
Nelore is a Bos indicus beef breed that is well-adapted to tropical environments and constitutes most of the world’s largest commercial cattle herd: the Brazilian bovine herd. Despite its significance, microbial genome recovery from ruminant microbiomes has largely excluded representatives from Brazilian Nelore cattle. To address this gap, this study presents a comprehensive dataset of microbial genomes recovered from the rumen and feces of 52 Brazilian Nelore bulls. A total of 1,526 non-redundant metagenome-assembled genomes (MAGs) were recovered from their gastrointestinal tract, with 497 ruminal and 486 fecal classified as high-quality. Phylogenetic analysis revealed that the bacterial MAGs fall into 12 phyla, with Firmicutes and Bacteroidota being the most predominant, while all archaeal MAGs belong to the genus Methanobrevibacter. The exploration of these microbial genomes will provide valuable insights into the metabolic potential and functional roles of individual microorganisms within host-microbiome interactions, contributing to a better understanding of the microbiome’s roles in bovine performance.
Similar content being viewed by others
Background & Summary
The gastrointestinal tract (GIT) of ruminants harbours a vast microbial ecosystem, termed the GIT microbiome, which plays critical roles in the digestive and immune systems of these animals1. The fermentation accomplished by the GIT microbiome influences production traits such as feed efficiency and methane emission2,3. This association between so many important processes and the GIT microbiome of ruminants indicates that its modulation could be a pivotal strategy to improve animal health and food quality while promoting more efficient and environmentally sustainable animal production systems4. But to achieve this, a comprehensive understanding of the composition, functionality, and interactions of the ruminant microbiomes is essential. Therefore, the present study aims to provide a comprehensive dataset that could serve as a foundation for more in-depth analyses on Nelore or ruminant microbiomes.
Despite the numerous advances regarding the study of ruminant microbiomes, there are still some gaps in our knowledge, with microbes whose characterization and role remain undefined or unknown5. Although this is strongly related to the inherent difficulties in cultivating certain microbes, the “Hungate1000” project recovered 410 bacterial and archaeal genomes from ruminant microbiomes through a combination of culturing and sequencing6. Nonetheless, culture-independent and reference-free approaches such as de novo assembly of shotgun metagenomic reads followed by binning into metagenome-assembled genomes (MAGs) have been developed7. This approach has significantly expanded the datasets of microbial genomes from diverse environmental niches, including the ruminant microbiomes5,7,8,9,10,11,12,13. Among the studies considering beef cattle animals, two stand out for having recovered 4,941 MAGs from the ruminal microbiome of Scottish cattle10 and 1,200 MAGs from the ruminal microbiome of African (Boran) cattle12. The successful recovery of these microbial genomes represents a significant accomplishment. Nevertheless, it’s important to emphasise that the GIT microbiomes and their associated functional potential can significantly differ due to factors such as diet, genetics, and the host animal’s environment3,5,14,15. Furthermore, most studies focus on rumen samples, even though significant taxonomic and functional variations are observed in the microbiomes distributed along the GIT3,5,15,16. Consequently, the current collection of microbial genomes obtained from microbiomes so far does not fully represent the diversity of the whole GIT ecosystem of bovines from different geographic locations, climates, and feeding regimes.
Nelore is a Bos indicus beef breed adapted to tropical environments and constitutes most of the biggest commercial herd in the world, the Brazilian bovine herd17. Despite its prominence, microbial genome studies focused on ruminant metagenomes have, until now, overlooked representatives from the Brazilian Nelore breed. In a recent work from our group, we analysed metagenomic data obtained from Nelore rumen and fecal microbiomes and unveiled significant associations between the bulls’ microbiomes and their diet and phenotypes3. However, these analyses only included the classification of metagenomic reads, which provides a broad perspective on the taxonomic profile and functional potential of the community, but falls short of exploring the microbiomes with a higher microbial resolution. Such limitations include the inability to assign functions to specific taxa, comprehend strain-specific genomic variations, and identify previously uncharacterized enzymes10,15,18.
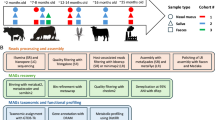
To reduce the underrepresentation of genomes from beef cattle microbiomes, we aimed to recover and characterise microbial genomes from the rumen and fecal samples of 52 Brazilian Nelore animals. A schematic diagram of the workflow followed in this study is presented in Fig. 1.

A schematic representation of the workflow applied to this study. Steps 1 and 2 were performed in our previous study3. Step 3 was applied to this study. The Nelore picture was taken by Gisele Rosso in 2023 and it belongs to Embrapa Southeast Livestock Multimedia: Image bank.
In this study, we single-assembled (assembly of each individual sample) and co-assembled (assembly of all samples from the same type) the metagenomic data from the ruminal content and fecal samples of 52 Brazilian Nelore steers3,19, producing over 60 million contigs totaling 63.9 gigabase pairs (Gbp). The bins obtained from the assemblies were aggregated and de-replicated at an average nucleotide identity (ANI) ≥99%, resulting in a total of the 1,526 GIT (789 ruminal and 737 fecal) non-redundant MAGs with completeness ≥50% and contamination ≤10%. Among these MAGs, 497 ruminal and 486 fecal were classified as high-quality (completeness ≥80%; contamination ≤10%; quality score ≥50) and were used for further analysis, while the remaining were classified as medium-quality (Fig. 2a).

Quality and metrics of MAGs recovered from the rumen and fecal microbiome of Nelore cattle. (a) Scatter plot illustrating the distribution of the recovered MAGs based on their completeness and contamination levels. Coloured dots represent the MAGs with completeness ≥80%, contamination ≤10% and quality score ≥50, considered high-quality MAGs. (b) Bar plots displaying the high-quality ruminal and fecal MAGs size with respect to the number of MAGs. (c) Box plots depicting the distribution of contig size, N50, and GC content among high-quality ruminal and fecal MAGs.
The genome size of the 983 High-Quality (HQ) MAGs ranges from 536 kilobases pairs (Kbp) to 5.8 megabases pairs (Mbp), with the majority falling within the range of 2–3 Mbp for HQ ruminal MAGs and 1.8–2.5 Mbp for HQ fecal MAGs (Fig. 2b). More than half of the HQ MAGs (n = 562) possessed less than 200 contigs (Fig. 2c). The majority of the HQ MAGs have N50 values ranging from 10 to 50 kb (Fig. 2c). GC content ranges from 0.2 to 0.6 in both ruminal and fecal HQ MAGs (Fig. 2c). Further information on the assemblies, bins, and HQ MAGs metrics can be found in Supplementary Tables 1, 2.
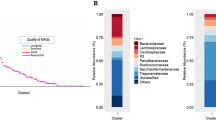
Taxonomic classification of the HQ MAGs revealed that they cover two microbial kingdoms, being 476 ruminal and 474 fecal MAGs assigned as Bacteria, whereas 21 ruminal and 12 fecal MAGs were assigned as Archaea. Complete taxonomic information can be found in Supplementary Table 3. The bacterial MAGs cover 12 known phyla (Fig. 3), mostly belonging to Firmicutes (n = 186 in rumen; n = 271 in feces) and Bacteroidota (n = 220 in rumen; n = 141 in feces). Among the Firmicutes, the majority belong to the class Clostridia (n = 153 in rumen; n = 227 in feces), followed by the classes Negativicutes (n = 10 in rumen; n = 26 in feces) and Bacilli (n = 23 in rumen;n = 18 in feces). Among the Bacteroidota, all MAGs belong to the class Bacteroidia and the order Bacteroidales (n = 220 in rumen; n = 141 in feces). Considering the MAGs classified as archaeal, all belong to the genus Methanobrevibacter (n = 21 in rumen; n = 12 in feces) (Fig. 4).


Phylogenetic tree illustrating the relationships among the 33 archaeal MAGs derived from Nelore’s microbiomes and closely related genomes. Methanosphaera sequences were used as outgroup. The tree was produced with GTDBtk30 and subsequently drawn using the ggtree32 package. All archaeal MAGs were assigned as Methanobrevibacter genus.
A predominance of MAGs assigned as Firmicutes and Bacteroidota was expected, as these are the most abundant phyla observed in the microbiomes of the animals studied3 as well as other ruminants1,6,15. Notably, taxa from these phyla have been associated with various factors of interest in animal production such as methane emission and feed efficiency2,3,20.
Similarly, a predominance of the genus Methanobrevibacter was expected since this is the most dominant archaeal genus within the microbiomes of ruminants2,10. Methanobrevibacter is a hydrogenotrophic methanogen, capable of using H2 to reduce CO2 into methane through the hydrogenotrophic pathway, the primary via of methane production in the rumen4.
Each of the 497 HQ ruminal MAGs had a taxonomic family assigned to it, consisting of 52 bacterial families and 1 archaeal family. 495 were classified to the genus level and 317 assigned to the species level. Regarding HQ fecal MAGs, all the 486 were classified up to the family level (46 bacterial families and 1 archaeal family). Of these, 470 were assigned to a genus and 215 were classified to the species level.
Notably, a fraction of these HQ MAGs was not assigned to a species (n = 180 in rumen and n = 271 in feces), while a smaller subset lacked both genus and species assignation (n = 2 in rumen and n = 16 in feces). This indicates a shortage of representatives for certain microbial groups and highlights the significance of studies aiming to recover genomes from microbiomes. Focused analyses should be conducted to explore the evolutionary relationships of these MAGs lacking complete taxonomy assignment.
Our study resulted in a comprehensive dataset of microbial genomes from the rumen and feces of Brazilian Nelore bulls. To the best of the authors’ knowledge, this marks the pioneering recovery of MAGs from this Bos indicus beef breed. The exploration of these microbial genomes will provide deep insights into the diverse roles of the microbiomes in methane emission, water footprint, feed efficiency, disease prevention, and overall bovine performance.
Methods
Metagenomic data
We processed and analysed ruminal and fecal metagenomes from 52 Nelore steers (Bos indicus), which comprises ~5.2 billion high-quality Illumina sequences (after the steps of trimming and filtering, and mapping against the host genome)19. The metagenomic data used in this study were previously published by our group3 and can be found under the BioProject ID PRJNA98774319. Briefly, total DNA was extracted from rumen content samples and fecal samples using the Quick-DNA™ Fecal/Soil Microbe Miniprep Kit (ZYMO Research Corp., Irvine, CA), metagenomic libraries were constructed with the Illumina DNA Prep Kit and sequenced on an Illumina NextSeq sequencer platform (ESALQ Genomics Center, Piracicaba, SP, Brazil) using the NextSeq P3 flowcell 300 cycles (Illumina). More information can be found in our previous study3, and in Supplementary Table 1.
The handling of the animals was conducted at the feedlot facility of “Embrapa Pecuária Sudeste” following Brazilian guidelines on animal welfare and approved by the Ethics Committee on the Use of Animals, College of Veterinary and Animal Science, São Paulo State University under protocol n° 8510190118 and EMBRAPA Livestock Science Ethics Committee on Animal Experimentation, São Carlos, São Paulo (Protocol No. 09/2016).
Metagenomic assembly, binning and MAGs recovery
The high-quality metagenomic sequences of each sample were individually assembled (single-assembled) and high-quality metagenomic sequences from all samples of the same sample type (rumen or feces) were co-assembled. The assemblies were performed with MEGAHIT v1.2.921 with options ‘–kmin-1pass–k-list 27,37,47,57,67,77,87–min-contig-len 1000’. Contigs from both single-metagenome assemblies and co-assemblies were grouped into draft genomes (bins) using three binning tools: MetaBAT2 v.2.1522 with option ‘–minContigLength 2000’, CONCOCT v.1.0.023 and MaxBin2 v.2.2.724, the later two with default parameters. The depth of coverage of each contig considered by the binning tools was calculated by mapping the raw reads back to their assemblies using BWA MEM v.0.7.1725 with default parameters, converting the mapping file to BAM format using Samtools v.1.1326. The contigs’ coverage was calculated using the script ‘jgi_summarize_bam_contig_depths’ for MetaBAT2 and MaxBin2 runs, and the script ‘concoct_coverage_table.py’ for CONCOCT run. The bins generated by these tools were integrated using the DAS tool27 with options ‘-l concoct,maxbin,metabat–search_engine diamond–write_bin_evals–write_bins’.
The bins were aggregated according to the sample type (rumen or feces) and then de-replicated using dRep v.3.2.228 with options ‘dereplicate -p 32 -comp 50 -con 10 -pa 0.95 -sa 0.99’, obtaining a set of 789 and 737 ruminal and fecal MAGs, respectively. In this process, only bins assessed by CheckM v1.1.329 as having medium quality (completeness ≥ 50% and contamination ≤ 10%) were considered for the de-replication workflow. After de-replication, the MAGs were filtered for completeness ≥ 80%, contamination ≤ 10% and quality score ≥ 50. Quality scores were defined as completeness − 5 × contamination, which only allows higher levels of contamination when the genome is predominantly complete8. This way, a total of 497 and 486 ruminal and fecal high-quality MAGs, respectively, were obtained and used for further analysis.
Taxonomic classification of the MAGs
To assign a taxonomy to each HQ MAG, GTDB-tk v2.3.2 was used with the GTDB database release 207 and options ‘classify_wf–full_tree–skip_ani_screen’. GTDB-tk generated separate phylogenetic trees for bacteria and archaea with the 983 HQ MAGs recovered and more than 60,000 genomes from the GTDB database. Taxonomy assignment of each MAG was based on its placement in the tree and its average nucleotide identity (ANI) to reference genomes. When rank assignments were considered ambiguous, the relative evolutionary divergence (RED) was used30. For better visualisation, a bacterial tree containing only the 950 bacterial MAGs was generated using GTDB-tk with option ‘infer’ and the sequence alignment previously generated by GTDB-tk. For the archaeal tree, the closest reference genomes to the archaeal MAGs were considered for the tree as well as Methanosphaera sequences, which were used as outgroup. GraPhlAn (Graphical Phylogenetic Analysis) v.1.1.431 was used to generate the figure of the tree with bacterial MAGs, and R package ggtree32 was used to generate the figure of the tree with archaeal MAGs.
For the submission of the high-quality MAGs to the National Center for Biotechnology Information (NCBI), the lowest taxonomic ranks assigned by GTDB for each MAG were retained if they were present in the NCBI taxonomy database; otherwise, they were replaced with the most appropriate taxonomic name recommended by NCBI. The best names tax names recommended by NCBI, NCBI Accession and links of each high-quality MAG are in Supplementary Table 3.
Mean coverage of the MAGs
Metagenomic reads from each sample were mapped to each MAG using Bowtie2 v2.5.333 with option ‘–no-unal’. SAMtools v1.19.234 was used to generate sorted BAM files with default parameters. Depth of coverage (mean) based on the sorted bam files generated with SAMtools was calculated using CoverM v0.7.0 (https://github.com/wwood/CoverM) with options ‘genome -m mean -m mean -min-read-aligned-percent 0.75 -min-read-percent-identity 0.95 -min-covered-fraction 0’.
Data Records
Raw reads used in this study19 are available at the National Center for Biotechnology Information (NCBI) under the BioProject Number PRJNA987743. The 983 high-quality Nelore MAGs generated in this study have been deposited in the same BioProject Number PRJNA98774335. Accession links for each high-quality MAG can be found in Supplementary Table 3.
Technical Validation
The metagenomic reads used in this study went through multiple steps of rigorous quality control, which included removing low-quality reads, adapters and host-associated sequences. These steps were performed using Trimmomatic and Bowtie2 as described in our previous study3. After assembly, only contigs greater than 1 Kbp were considered, as small contigs tend to carry less compositional signatures, which can bias the binning step. The quality of the recovered MAGs was assessed using CheckM and only those with completeness ≥80%, contamination ≤10% and quality score ≥50 were used in the downstream analyses. These metrics are similar to those used in previous studies focused on recovering MAGs from beef cattle microbiomes10,12,15.
Code availability
The present study did not use custom scripts to generate the dataset. The parameters and versions of all the bioinformatics tools used for the analysis are described in the Methods section. The code used to run each of the tools is publicly available at Github (https://github.com/lconteville/Nelore_MAGs).
References
Welch, C. B., Ryman, V. E., Pringle, T. D. & Lourenco, J. M. Utilizing the Gastrointestinal Microbiota to Modulate Cattle Health through the Microbiome-Gut-Organ Axes. Microorganisms 10, 1391 (2022).
Greening, C. et al. Diverse hydrogen production and consumption pathways influence methane production in ruminants. ISME J. 13, 2617–2632 (2019).
Conteville, L. C. et al. Rumen and fecal microbiomes are related to diet and production traits in Bos indicus beef cattle. Front. Microbiol. 14, (2023).
Huws, S. A. et al. Addressing Global Ruminant Agricultural Challenges Through Understanding the Rumen Microbiome: Past, Present, and Future. Front. Microbiol. 9, 2161 (2018).
Xie, F. et al. An integrated gene catalog and over 10,000 metagenome-assembled genomes from the gastrointestinal microbiome of ruminants. Microbiome 9, 137 (2021).
Seshadri, R. et al. Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection. Nat. Biotechnol. 36, 359–367 (2018).
Chen, L.-X., Anantharaman, K., Shaiber, A., Eren, A. M. & Banfield, J. F. Accurate and complete genomes from metagenomes. Genome Res. 30, 315–333 (2020).
Parks, D. H. et al. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2, 1533–1542 (2017).
Stewart, R. D. et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 9, 870 (2018).
Stewart, R. D. et al. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 37, 953–961 (2019).
Li, J. et al. A catalog of microbial genes from the bovine rumen unveils a specialized and diverse biomass-degrading environment. GigaScience 9, giaa057 (2020).
Wilkinson, T. et al. 1200 high-quality metagenome-assembled genomes from the rumen of African cattle and their relevance in the context of sub-optimal feeding. Genome Biol. 21, 229 (2020).
Nayfach, S. et al. A genomic catalog of Earth’s microbiomes. Nat. Biotechnol. 39, 499–509 (2021).
Li, F. et al. Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle. Microbiome 7, 92 (2019).
Lin, L., Lai, Z., Zhang, J., Zhu, W. & Mao, S. The gastrointestinal microbiome in dairy cattle is constrained by the deterministic driver of the region and the modified effect of diet. Microbiome 11, 10 (2023).
Lopes, D. R. G. et al. The Bacterial and Fungal Microbiota of Nelore Steers Is Dynamic Across the Gastrointestinal Tract and Its Fecal-Associated Microbiota Is Correlated to Feed Efficiency. Front. Microbiol. 10, 1263 (2019).
Lima, P. R. M., Peripolli, V., da Silva, L. O. C. & McManus, C. Spatial distribution of genetic values of Nelore breed in Brazil. Livest. Sci. 250, 104599 (2021).
Zeng, S. et al. A compendium of 32,277 metagenome-assembled genomes and over 80 million genes from the early-life human gut microbiome. Nat. Commun. 13, 5139 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP445878 (2023).
Andrade, B. G. N. et al. Stool and Ruminal Microbiome Components Associated With Methane Emission and Feed Efficiency in Nelore Beef Cattle. Front. Genet. 13, 812828 (2022).
Li, D. et al. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods San Diego Calif 102, 3–11 (2016).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Wu, Y.-W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinforma. Oxf. Engl. 32, 605–607 (2016).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl. 26, 589–595 (2010).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinforma. Oxf. Engl. 25, 2078–2079 (2009).
Sieber, C. M. K. et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3, 836–843 (2018).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinforma. Oxf. Engl. btz848 https://doi.org/10.1093/bioinformatics/btz848 (2019).
Asnicar, F., Weingart, G., Tickle, T. L., Huttenhower, C. & Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3, e1029 (2015).
Yu, G., Smith, D. K., Zhu, H., Guan, Y. & Lam, T. T.-Y. ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 (2017).
Langmead, B. & Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9, 357–359 (2012).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience. 10(2), giab008 (2021).
NCBI BioProject. https://identifiers.org/ncbi/bioproject:PRJNA987743 (2024).
Acknowledgements
The authors thank Leandro Carrijo Cintra for the support with the Embrapa Agricultural Digital (CNPTIA) Server. This research was supported by a postdoctoral fellowship to LCC from Fundação de Amparo à Pesquisa do Estado de São Paulo fellowship (FAPESP, 2020/15565–7), a productivity fellowship to LLC and LR from Conselho Nacional de Desenvolvimento Científico e Tecnológico, and funding from Fundação de Amparo à Pesquisa do Estado de São Paulo Thematic Grant (FAPESP, grant number 2019/04089–2), and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, grant number 428153/2018).
Author information
Authors and Affiliations
Contributions
L.C.C.: Conceptualization, Formal Analysis, Methodology, Writing – original draft, Investigation, Validation, Visualization. J.V.S.: Writing – original draft, Investigation. B.G.N.A.: Conceptualization, Writing – review & editing, Investigation. L.L.C.: Supervision, Writing – review & editing, Funding acquisition, Project administration. J.C.P.P.: animal experiment design and execution, Supervision, Writing – review & editing, Data curation. L.C.A.R.: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Conteville, L.C., Silva, J.V.d., Andrade, B.G.N. et al. Recovery of metagenome-assembled genomes from the rumen and fecal microbiomes of Bos indicus beef cattle. Sci Data 11, 1385 (2024). https://doi.org/10.1038/s41597-024-04271-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-04271-3
