
Abstract
The beet webworm, Loxostege sticticalis Linnaeus (Lepidoptera, Pyralidae), is a major pest in agriculture and livestock production. However, the L. sticticalis genome has not yet been sequenced, limiting exploration of its biological features and population genetics. In this study, the genome of L. sticticalis was sequenced on the Illumina Novaseq. 6000 and PacBio Sequel II platforms, and chromosome conformation capture (Hi-C) methods were used to generate the high-quality chromosome-level genome, assessed at 98.7% by the Benchmarking Universal Single-Copy Orthologs (BUSCO) tool. The L. sticticalis genome showed an assembly of 485.9 Mb with a contig N50 of 16.4 Mb, a scaffold N50 of 16.6 Mb, and a GC content of 37.85%, with over 98.67% of the assembled bases located on 31 chromosomes. Repeat sequences accounted for 41.71% of the genome and 15 913 protein-coding genes were identified. Comparison of the genome of L. sticticalis with other closely related species indicated high chromosomal synteny. The sequencing of this genome contributes to research on the genetics and evolution of the Lepidoptera.
Similar content being viewed by others


Background & Summary
The beet webworm, Loxostege sticticalis Linnaeus (Lepidoptera, Pyralidae), is a major agricultural pest in North America, Europe, and Asia1,2. Outbreaks of this pest occur over a wide region (108-118° E, 37-35° N) of China, with three periods of outbreaks reported over the last 70 years. Since 2018, L. sticticalis has entered the fourth outbreak period in China, resulting in serious ecological and economic losses, especially in the northern part of China3,4.
In 2020, the Ministry of Agriculture and Rural Affairs listed L. sticticalis in the List of National Class I of Crop Diseases and Insect Pests. In China, L. sticticalis generations increase from 1 to 4 with decreasing latitude and altitude. However, the main damage generations of L. sticticalis are the first and second generations5. L. sticticalis is classified as a facultative migrant pest and shows significant ability to crawl and migrate. The fourth and fifth instar larval stages are the most destructive as they usually consume all available leaves within an area6. L. sticticalis can damage more than 200 species belonging to 35 families of host plants, including Glycine max, Helianthus annuus, Chenopodium album, Beta vulgaris, and Zea mays, thus posing a major threat to the production of crops, such as grains, oilseeds, and pastures (Fig. 1)3.

Life cycle of Loxostege sticticalis and its damage on maize. (a) Diferent developmental stages of L. sticticalis. (b) The symptom of maize leaves damaged by L. sticticalis.
Currently, a variety of chemical pesticides have been used for widespread control of L. sticticalis, however, this management strategy can lead to insecticide resistance increase and negatively impact the agro-ecosystem. Genomic analysis can be helpful for the development of integrated pest-management approaches, as has been demonstrated7,8. To date, the genomes of several typical migrant pests have been sequenced and published, such as Mythimna separate9, Helicoverpa armigera10, and Spodoptera frugiperda11. This information leads to an understanding of molecular and genetic mechanisms for pest control. However, the genome of L. sticticalis, a member of the Class I of Crop Diseases and Insect Pests, has not yet been sequenced. Therefore, to promote the development of innovative management strategies for controlling L. sticticalis, chromosome-level genome is necessary and, in this study, we present the chromosome-level genome of L. sticticalis. This provides a resource for studies on L. sticticalis, as well as novel insights into the evolution and ecology of migrant pests.
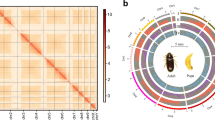
In this study, three male pupae were used for the whole-genome assembly. Firstly, the genome of L. sticticalis was surveyed before sequencing, finding that the size of the genome was an estimated 1.9 Gb, with a repetitive sequence content of 67.87% and a heterozygosity rate of 0.31% (Fig. 2a). Then, we generated 31.4 Gb of CCS data using the PacBio SequeII platform and assembled the genome, with an overall content of 485.9 Mb and a repetitive sequence content of 41.71%, consisting of 118 contigs with an scaffold N50 length of 16.4 Mb, associated with 31 chromosomes (Table 1, Fig. 2b,c). The L. sticticalis genome size is inconsistency of the results of survey and third-generation sequencing, which is due to the high heterozygosity of insect12. In addition, the Benchmarking Universal Single-Copy Orthologs (BUSCO) (v5.2.1, odb10) tool was used to assess the completeness of the L. sticticalis genome, yielding a value of 98.7% with a signal copy value of 97.7%, and duplicated copy rate of 1.0% (Table 2), indicating the good quality of the genome assembly and annotation13.

Overview of the genomic landscape of the beet webworm, Loxostege sticticalis. (a) Characteristics of the Illumina short-read sequencing of the L. sticticalis genome. (b) Circle genomic landscape of L. sticticalis. The circles from the outside to the inside represent chromosome sequence, gene density, GC content, and repeat sequence content, respectively. The middle lines of the circles indicate genes showing collinearity. (c) Hi-C interactive heatmap of L. sticticalis.
Meanwhile, 41.71% of the genome was annotated as repeat sequences (Supplementary Table 1), while DNA transposons, long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), and long terminal repeats (LTRs) accounted for 4.4, 9.69, 0.79 and 6.44%, respectively, of the whole L. sticticalis genome. In addition, 11.52% repeat sequences within the whole genome were not classified. In terms of protein-coding genes, a total of 17 431 genes were predicted and annotated in the assembled genome (Supplementary Table 2). Of these, 17 113 (98.17%) of the encoded proteins were annotated using different databases, including NR, Swiss-Prot, Gene Ontology (GO), Clusters of Orthologous Genes (COG), Eukaryotic Orthologous Groups (KOG), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Pfam (Supplementary Table 3). Moreover, different types of non-coding RNA were identified, including 15 930 tRNAs with tRNASCAN, 90 miRNAs, 130 rRNAs, and 97 snRNAs (Supplementary Table 4, 5, 6).
Orthologous genes were identified by comparison of L. sticticalis and 15 other fifty lepidopteran insects. In total, 11 719 gene families were identified, within which 1 867 single-copy and 4 065 multiple-copy orthologs were found (Supplementary Table 7). The phylogenetic relationships and estimated times of species divergence were analyzed using the protein sequences of the single-copy orthologous genes. The results indicated that L. sticticalis and Ostrinia furnacalis shared the last common ancestor approximately 42.8 million years ago (Fig. 3).

Phylogenetic tree and gene orthology of 16 lepidopteran insect genomes. The maximum-likelihood phylogenetic tree was constructed with 1000 bootstrap replicates. The blue node indicates the correction point, while light blue indicates the 95% confidence interval. The number adjacent to the node represents the time of divergence in million years ago. The numbers on the branch represent expansions (red) or contractions (blue) of the gene families.
The expansion and contraction of L. sticticalis gene families, as well as those of related species, were analyzed using CAFÉ (v4.2.1) software. The results showed expansion of 785 gene families in L. sticticalis, and 688 families have contracted (Fig. 3). Meanwhile, GO enrichment analysis showed that these 785 expanded gene families in L. sticticalis genome were mainly involved in “integral component of membrane (GO:0016021)”, “RNA-directed DNA polymerase activity (GO:0003964)”, and “DNA integration (GO: 0015074)”. The 688 contracted families were mainly involved in “extracellular region (GO:0005576)”, “RNA-directed DNA polymerase activity (GO:0003964)”, and “DNA integration (GO:0015074)” (Fig. 4, Supplementary Fig. 1, 2).

GO enrichment analysis of Loxostege sticticalis gene families showing expansion (a) and contraction (b).
Four lepidopteran insects were selected for syntenic analysis. Generally, the Lepidoptera shared high chromosomal synteny, although several fusion events were detected between L. sticticalis and other Lepidopteran species (Fig. 5), specifically, between Cydia pomonella chromosome 1 and chromosomes 1 and 3 of L. sticticalis and C. pomonella chromosome 2 with chromosomes 11 and 25 of L. sticticalis. A number of intrachromosomal inversions and other local rearrangements were also detected.

Synteny analysis between four species, Loxostege sticticalis, Cydia pomonella, Chilo suppressalis and Plutella xylostella. Each rounded rectangle represents a chromosome, and the line in the middle indicates a collinear block.
For phytophagous insects, detoxification and chemosensation abilities are both essential for selecting and locating host plants14,15,16. These feeding preferences can be reflected at the level of the genome, as previously described13,15,17. In this study, associations were also observed between gene numbers and host ranges (Fig. 6). For instance, in terms of detoxification-related genes, the numbers of genes in the cytochrome P450 (P450) families were observed to increase sequentially in monophagous, oligophagous, and polyphagous insects. Furthermore, greater numbers of chemosensory-related genes have been observed in insects with broader host ranges13. In L. sticticalis, 81 gustatory receptors (GRs) were identified, which is higher than the number in monophagous and polyphagous insects The results indicate that the feeding preference of the insect is correlated with the number of genes involved in detoxification and chemosensing. Taken together, a high-quality chromosome-level genome of L. sticticalis was assembled, providing a valuable genomic resource for the further understanding of insect olfactory, evolutionary, and feeding preferences. Moreover, this genomic resource provides a reference for the implementation of integrated pest-management strategies for migratory insects.

Distribution of detoxification and chemosensory genes in 18 Lepidopteran insects. The numbers in the cells indicate the size of the corresponding gene family for each species. A darker background color in the cells indicates that more genes were encoded in the corresponding species.
Methods
Sample collection and DNA extraction
The first generation of L. sticticalis was collected in Huhhot, Inner Mongolia Province, China (40°82′ N, 111°71′ E). The larvae were reared under laboratory conditions at a constant temperature of 22 ± 1°C, photoperiod of 16:8 (L:D), and relative humidity of 75 ± 5%, with feeding with fresh Chenopodium album. The fifth (last) instar larvae were transferred to a box for pupation, containing clean sandy soil with 15% humidity. The food of the adults was supplemented with a 5% honey solution. Overall, 1 male and 1 female collected in the field were reared in the lab, and the pupae of the third generation were used for genome sequencing. Three male pupae were used for high-quality genomic DNA extraction, using the sodium dodecyl sulfate (SDS) method18. The RNA contaminants were removed by RNase A. The DNA quality, concentration, and integrity were assessed using 0.5% agarose gel electrophoresis (AGE), a Nanodrop spectrophotometer (Thermo Fisher, Waltham, MA, USA; NANODROP2000), and Qubit fluorometry (Invitrogen, QubitTM3Flurometer).
Library construction and genome sequencing
For short-read sequencing, a paired-end library was constructed with an insert size of 350 bp, after which 150 bp paired-end reads (PE 150) were used for whole-genome sequencing on the Illumina NovaSeq. 6000 platform (Illumina, San Diego, CA, USA), following the provided instructions. For long-read sequencing, the genomic DNA was sheared into 15 kb fragments by g-TUBE (Covaris), and a SMRTbell library was constructed using a SMRTbell Express Template Prep kit 2.0 (PacBio, 100-938-900). The size distribution and concentration of the library were assessed using a FEMTO Pulse automated pulsed-field capillary electrophoresis instrument (Agilent Technologies, Wilmington, DE, USA) and a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). The genomic DNA library was purified with 1X AMPure PB beads (Pacbio, Menlo Park, CA, USA) and sequenced on both the Illumina and PacBio Sequel II platforms, following the accompanying instructions. A Sage ELF system (Sage Science, Beverly, MA, USA) was used to select the SMRTbell range of 15 to 18 kb, and the library was then purified using 1X AMPure PB beads. The final SMRTbell library size and quantity were assessed using the FEMTO Pulse and the Qubit dsDNA HS reagents assay kit. The sequencing primer and Sequel II DNA Polymerase of the library were annealed and bound. SMRT sequencing was performed using a single 8 M SMRT Cell on the Sequel II System with the Sequel II Sequencing Kit and 1800-minute movies by Biomarker Technologies Co., Ltd. (Qingdao, China).
Estimation of genome features
The fastp tool (version 0.20.0, https://github.com/OpenGene/fastp) (Chen et al., 2018) was used for quality filtration of the short-reads from the Illumina sequences using the parameters -q 10 -u 50 -y -g -Y 10 -e 20 -l 100 -b 150 -B 150. First, the adaptors were removed from the sequenced reads. Second, read pairs were excluded if any one end had an average quality of less than 20. Third, the ends of the reads were trimmed if the average quality was lower than 20 in the sliding window size of 5 bp. Finally, read pairs with any ends shorter than 75 bp were removed. The quality-filtered reads were then used for genome size estimation. The genome size, heterozygosity, and the repeat contents were estimated using the filtered reads of 21-mer with Jellyfish software (-h 1000000000)19 and the genome characteristics were determined using Genomescope 2.0 (-k 21 -p 6 -m 100000)20. The completeness of the genomic annotation was evaluated by Benchmarking Universal Single-Copy Orthologs (BUSCO, version: 5.2.1, odb10) software.
Genome assembly from CCS data and anchor contigs
High-accuracy CCS data were assembled using hifiasm (v0.16.1, https://github.com/chhylp123/hifiasm) software to obtain the genome sequences. Paired reads with mates mapped to a different contig were used for the Hi-C-associated scaffolding. The Hi-C technology was used to help anchor contigs. The contigs were clustered using ALLHIC (v0.9.8) software21 to determine the closeness of associations between contigs. The interactions between two contigs were converted into the specified binary files (Hi-C files) using juciertools (v3.0, https://github.com/aidenlab/JuicerTools) software. The sequenced and oriented contigs were then manually corrected using Juciebox (v2.15.07)22 to obtain the chromosome-level assembly results and were mapped to the polished L. sticticalis genome using BWA (bwa-0.7.17) with default parameters. Self-ligation, non-ligation, and other invalid reads, such as Start NearRsite, PCR amplification, random break, Large Smal lFragments, and Extreme Fragments, were filtered.
Genome annotation
De novo and homology-based methods were used to identify repetitive sequences in the L. sticaticalis genome. First, RepeatModeler (v2.0.1, http://www.repeatmasker.org/RepeatModeler/)23 was used to construct a de novo repeat library, and the predicted results were merged by the RepBase database (http://www.girinst.org/repbase). Second, RepeatMasker (v4.1.0, http://www.repeatmasker.org) was used to predict the repetitive sequences of the L. sticticalis genome, and theRepeatProteinMask tool in RepeatMasker was used to predict the repetitive sequences. The two results were then integrated.
Three methods were used to predict mRNA, including de novo prediction (Augustus v3.3.3, https://github.com/Gaius-Augustus/Augustus and GlimmerHMM v3.0.424, homology searches (exonerate v2.4.0, https://github.com/nathanweeks/exonerate/ and GeMoMa v1.6.425, and transcript prediction, which the the RNA-seq transcripts were re-constructed through StringTie v2.1.3 (TransDecoder v5.1.0)26, followed by the use of TransDecoder v5.1.0 (https://github.com/TransDecoder/TransDecoder) to predict protein-coding genes. The obtained multiple datasets were integrated using EVidenceModeler (v1.1.1)27, and finally the integrated data were updated, UTR regions were added, and new transcripts were identified using PASA (v2.5.2, https://github.com/PASApipeline/PASApipeline). BLAST v2.10.1+28 was used to compare the the longest nucleic acid transcript sequences to six database, namely, NR, Swiss-Prot, GO, COG, KOG and KEGG. The protein sequences were analyzed in the Pfam database by HMMER V3.2.129 software.
For annotation of non-coding RNA, barrnap 0.9 (https://github.com/tseemann/barrnap) and tRNASCAN v2.0.030 were used to predict ribosomal RNA (rRNA) and transfer RNA (tRNA), respectively. The Rfam database was used to predict non-coding RNA using infernal 1.1.3 (https://github.com/EddyRivasLab/infernal).
Gene family orthology and phylogenetic analyses
For analysis of orthologous genes in the gene families, 16 lepidopteran species, including L. sticticalis, Bombyx mori31, Ostrinia furnacalis32, Helicoverpa armigera10, Spodoptera frugiperda11, Cydia pomonella7, Mythimna separate9, Mythimna loreyi9, Manduca sexta33, Heliothis virescens34, Spodoptera exigua35, Spodoptera litura36, Chilo suppressalis37, Cnaphalocrocis exigua8, Trichoplusia ni38, and Plutella xylostella39 were selected. The longest amino sequences were used for ortholog identification by OrthoFinder (2.3.5) with clustering by diamond (v2.0.6.144). The signal-copy orthologous genes from the OrthoFinder results were aligned using MAFFT (v7.427), and phylogenetic trees were constructed using RAxML (8.2.12) with 1000 bootstrap replicates and model PROTGAMMAJTT. The phylogenetic tree with divergence times was constructed using MCMCTREE (v4.9 h).
Analysis of gene family expansion and contraction
The results of the gene clustering were filtered to remove families containing >100 genes in one species. Analysis of gene family expansion and contraction was performed with CAFÉ (v4.2.1) with a P-value threshold <0.05 as the cut-off. In addition, genes in families showing expansion and contraction were compared using the GO database by BLAST (v2.10.1+) for analysis of the gene function.
Chromosome synteny analysis
For the chromosome synteny analysis, four lepidopterans species, namely, L. sticticalis, Cydia pomonella7, Chilo suppressalis37 and Plutella xylostella39, were selected. The Multiple Collinearity Scan toolkit (MCScanX) was used to identify collinearity.
Gene family identification
To investigate the reasons underlying the feeding habits of L. sticticalis at the genomic level, detoxification- and chemosensing-associated genes from 18 lepidopteran insects (16 species in the part of “Gene family orthology and phylogenetic analyses”, Leguminivora glycinivorella40 and Danaus plexippus41) were compared using BLASTP (E < 10−5) and TBLASTN (E < 10−5). Specifically, detoxification-associated genes, including cytochrome P450 (P450), ATP-binding cassette (ABC), carboxyl/cholinesterases (CCE), UDP-glycosyltransferases (UGT), and glutathione-S-transferase (GST), as well as chemosensing-associated genes, including odorant binding proteins (OBPs), chemosensory proteins (CSPs), odorant receptors (ORs), ionotropic receptors (IRs), and gustatory receptors (GRs) were analyzed.
Data Records
The NCBI BioProject number for the data reported in this paper is PRJNA111849242. The cleansequencing data for short read, Hi-C and HiFi have been deposited in the NCBI Sequence Read Archive with accession number SRR2936623243, SRR2936623144, SRR2936623045, respectively. The chromosomal-level genome assembly file was deposited in the NCBI GenBank with accession number JBEDNZ00000000046.
Technical Validation
The chromosome-level genome of the L. sticticalis assembly was 485.9 Mb with a scaffold N50 length of 16.6 Mb. Assessment of the completeness of the genome was performed using BUSCO (version: 5.2.1, odb10). The results showed that more than 98% of BUSCO genes were identified in the genome assembly (Table 2), indicating a high level of completeness of the L. sticticalis genome assembly. Moreover, the Hi-C heatmap showed obvious grouping, and the intensity of the interaction was higher than that of the off-diagonal position, indicating a high degree of interaction between neighboring sequences in the chromosomal results of the Hi-C assembly, while the interaction signals between non-adjacent sequences were weak, consistent with the principle of Hi-C auxiliary assembly, indicating better genomic anchoring effect. The genome of L. sticticalis was divided into 31 groups, indicating 31 chromosomes (Fig. 2c). Thus, the chromosome-level genome of L. sticticalis was of high quality.
Code availability
All bioinformatic tools and software for data analysis in this study were used according to the manuals, and the versions and code/parameters of the software have been included in the Methods section. No custom code was used.
References
Kong, H. et al. Molecular characterization of a lysozyme gene and its altered expression profile in crowded beet webworm (Loxostege sticticalis). PLoS One 11, e0161384 (2016).
Shakhmaev, R. N. et al. Stereoselective synthesis of 11(E)-tetradecen-1-yl acetate-sex pheromone of sod webworm (Loxostege sticticalis). Russ. J. Gen. Chem. 79, 1171–1174 (2009).
Feng, H. et al. Spring migration and summer dispersal of Loxostege sticticalis (Lepidoptera: Pyralidae) and other insects observed with radar in Northern China. Environ. Entomol. 33, 1253–1265 (2004).
Jiang, X. F. et al. The fourth occurrence cycle of the beet webworm Loxostege sticticalis may be coming in China. Plant Protec. 45, 79–81 (2019). in Chinese.
Jiang, X. F. et al. Diapause termination, post-diapause development and reproduction in the beet webworm, Loxostege sticticalis (Lepidoptera: Pyralidae). J. Insect. Physiol. 56, 1325–1331 (2010).
Wen, M. et al. A herbivore-induced plant volatile of the host plant acts as a collective foraging signal to the larvae of the meadow moth, Loxostege sticticalis (Lepidoptera: Pyralidae). J. Insect. Physiol. 118, 103941 (2019).
Wan, F. et al. A chromosome-level genome assembly of Cydia pomonella provides insights into chemical ecology and insecticide resistance. Nat. Commun. 10, 4237 (2019).
Xu, H. et al. Chromosome‐level genome assembly of an agricultural pest, the rice leaffolder Cnaphalocrocis exigua (Crambidae, Lepidoptera). Mol. Ecol. Resour. 22, 307–318 (2021).
Zhao, H. et al. Chromosome-level genomes of two armyworms, Mythimna separata and Mythimna loreyi, provide insights into the biosynthesis and reception of sex pheromones. Mol. Ecol. Resour. 23, 1423–1441 (2023).
Zhang, J. et al. Population genomics provides insights into lineage divergence and local adaptation within the cotton bollworm. Mol. Ecol. Resour. 22, 1875–1891 (2022).
Zhang, L. et al. Genetic structure and insecticide resistance characteristics of fall armyworm populations invading China. Mol. Ecol. Resour. 20, 1682–1696 (2020).
Elliott, T. A. et al. What’s in a genome? The C-value enigma and the evolution of eukaryotic genome content. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370. (2015).
Zhou, S. et al. A chromosome-level genome assembly of yellow stem borer (Scirpophaga incertulas). Sci. Data 11, 279 (2024).
Crespo, J. G. A review of chemosensation and related behavior in aquatic insects. J. Insect Sci. 11, 1–39 (2011).
Rane, R. V. et al. Detoxifying enzyme complements and host use phenotypes in 160 insect species. Curr. Opin. Insect Sci. 31, 131–138 (2019).
Renou, M. et al. Insect olfactory communication in a complex and changing world. Curr. Opin. Insect Sci. 42, 1–7 (2020).
Edger, P. P. et al. The butterfly plant arms-race escalated by gene and genome duplications. Proc. Natl. Acad. Sci. USA 112, 8362–8366 (2015).
Wang, Y. S. et al. Comparative analysis of eight DNA extraction methods for molecular research in mealybugs. PloS One 14, e0226818 (2019).
Marcais, G. et al. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Ranallo-Benavidez, T. R. et al. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Zhang, X. et al. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Robinson, J. T. et al. Juicebox.js provides a cloud-based visualization system for Hi-C data. Cell Syst. 6, 256–258 (2018).
Flynn, J. M. et al. Repeat Modeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA. 117, 9451–9457 (2020).
Majoros, W. H. et al. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Keilwagen, J. et al. GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol. Biol. 1962, 161–177 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Altschul, S. F. et al. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Eddy, S. R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 23, 205–211 (2009).
Chan, P. P. et al. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096 (2021).
Lu, F. et al. SilkDB 3.0: visualizing and exploring multiple levels of data for silkworm. Nucleic Acids Res. 48, D1 (2020).
Fang, G. et al. The draft genome of the Asian corn borer yields insights into ecological adaptation of a devastating maize pest. Insect Biochem. Mol. Biol. 138, 103638 (2021).
Gershman, A. et al. De novo genome assembly of the tobacco hornworm moth (Manduca sexta). G3 (Bethesda). 11, jkaa047 (2021).
Fritz, M. L. et al. Contemporary evolution of a Lepidopteran species, Heliothis virescens, in response to modern agricultural practices. Mol. Ecol. 27, 167–181 (2017).
Simon, S. et al. Genome and transcriptome analysis of the beet armyworm Spodoptera exigua reveals targets for pest control. G3 (Bethesda). 11, jkab311 (2021).
Cheng, T. et al. Genomic adaptation to polyphagy and insecticides in a major East Asian noctuid pest. Nat. Ecol. Evol. 1, 1747–1756 (2017).
Ma, W. et al. A chromosome-level genome assembly reveals the genetic basis of cold tolerance in a notorious rice insect pest, Chilo suppressalis. Mol. Ecol. Resour. 20, 268–282 (2019).
Chen, W. et al. A high‐quality chromosome‐level genome assembly of a generalist herbivore, Trichoplusia ni. Mol. Ecol. Resour. 19, 485–496 (2019).
Jouraku, A. et al. KONAGAbase: A genomic and transcriptomic database for the diamondback moth, Plutella xylostella. BMC Genom. 14, 464 (2013).
Wang, Y. et al. A chromosome-level genome assembly of the soybean pod borer: insights into larval transcriptional response to transgenic soybean expressing the pesticidal Cry1Ac protein. BMC Genom. 25, 355 (2024).
Mongue, A. J. et al. Neo-sex chromosomes in the monarch butterfly, Danaus plexippus. G3 (Bethesda) 7, 3281–3294 (2017).
NCBI National Genomics Data Center (NGDC) database https://identifiers.org/ncbi/bioproject:PRJNA1118492 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29366232 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29366231 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29366230 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBEDNZ000000000.1 (2024).
Acknowledgements
This work was supported by National Key Research and Development Program of China (2022YFD1400600), Central Government to Guide Local Science and Technology Development Fund Projects of China (2021ZY0041), Natural Science Foundation of Inner Mongolia (2022MS03012), 2021 High Level Talents Project of Inner Mongolia (2022NMRC010), the Special Fund for Basic Scientific Research Business of Central Public Welfare Scientific Research Institutes (1610332022010), the Inner Mongolia Science and Technology Plan Project (No.2021GG0396) and Natural Science Foundation of Inner Mongolia Autonomous Region of china (No.2021MS03075).
Author information
Authors and Affiliations
Contributions
K.J.L. and S.J.G. conceived the project; Y.M.S., Y.W., L.W.G., N.Y.L. and Q.Z. prepared the samples for sequencing; Z.Y. and P.H.B. performed the experiments; P.H.B., Y.Y.W., L.X., Y.M.S., Y.W., and L.W.G. performed functional analysis; Z.Y., Y.Y.L., H.B.H., L.X. and L.B.X. performed the bioinformatic analyses; H.B.H., Q.Z., Y.Y.W., and N.Y.L. performed the figure draw; K.J.L., L.B.X., and S.J.G. evaluated the results; Z.Y. and Y.Y.L. wrote and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Y., Li, Y., Shan, Y. et al. A chromosome-level genome assembly of beet webworm, Loxostege sticticalis Linnaeus (Lepidoptera: Pyralidae). Sci Data 12, 869 (2025). https://doi.org/10.1038/s41597-025-04371-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04371-8
