
Abstract
Goosegrass, belonging to the genus Eleusine within the Chloridoideae subfamily, is often one of the problematic weeds with strong invasiveness, competing with crops for essential survival resources. Although a chromosome-level genome assembly of E. indica from China was published last year, the present research focuses on a population of E. indica native to South Korea. Considering the high genetic variability among wild E. indica populations, constructing multi-reference genomes from geographically distinct populations is crucial for comprehensive weed management strategies. In this study, we sequenced and assembled the whole genome using PacBio long read and Illumina short read sequencing platforms. We then combined Pore-C sequencing technology to successfully anchor 255 contigs to nine pseudochromosomes. The chromosome-level genome assembly showed a high level of completeness with a 97% score according to BUSCO analysis results. Repetitive sequences accounted for 97% of the genome assembly, and 26,836 protein-coding genes were predicted. The high-quality genome assembly of E. indica will serve as a valuable genetic resource to enhance our understanding of weed control research.
Similar content being viewed by others


Background & Summary
Eleusine indica, commonly known as goosegrass, is a globally distributed weed that competes with crops for essential resources in modern agriculture1. Known for its invasive nature and strong survival strategies2, this plant is an annual with a chromosome number of 2n = 2x = 18 and is a self-pollinating diploid species3. It adapts well to various habitats, including tropical and subtropical regions4, and shows high tolerance to extreme conditions such as high temperatures, drought, and low mowing5. E. indica, commonly found in rice production areas6, produces about 40,000 seeds per plant and has a high tillering ability, causing significant crop yield losses7. To understand the adaptive strategies and evolutionary processes of the Eleusine genus, the development of high-quality reference genomes is essential.
Although a chromosome-level genome assembly of E. indica from China was completed and published last year8, this study focuses on a population of E. indica native to South Korea. Given the high genetic variability in wild E. indica populations9, constructing multi-reference genomes with geographically distinct populations is crucial. The Korean E. indica population represents a unique genetic pool that may exhibit significant differences due to local environmental pressures and adaptation mechanisms. Therefore, the aim of this study is to provide a comprehensive chromosome-level genome assembly of E. indica from South Korea, offering insights into the genetic diversity and potential adaptive traits specific to this population.
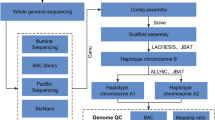
In this study, we constructed a high-quality chromosome-level genome assembly of E. indica using a combination of PacBio long-read sequencing, Illumina short-read sequencing, and Pore-C sequencing data. The assembled genome size is approximately 478 Mb, with 98.48% of the genome successfully anchored to nine pseudochromosomes. Of the assembled genome, 59.76% consists of repeat sequences, of which 39.93% are transposable elements containing long terminal repeats (LTRs). Additionally, the genome includes 26,836 protein-coding genes. These results indicate the high quality of the E. indica genome assembly, which will contribute to a broader understanding of the genomic landscape of E. indica. This underscores the importance of studying diverse populations to fully comprehend the genetic complexity and evolutionary dynamics of E. indica. By providing a high-quality reference genome for the Korean E. indica population, our study establishes a foundational resource that will contribute to a future pan-genome project for E. indica, enabling a comprehensive understanding of genomic diversity across populations. The E. indica genome assembly presented here serves as a valuable genetic resource for improving crop resilience and advancing effective weed management strategies.
Methods
Sample collection, genomic DNA and RNA extraction
The seeds of E. indica were collected from the area around Geumnung, Jeju Province, South Korea (33′23″18.72,126′13″37.02), and are stored in the Plant Computational Genomics Laboratory at the College of Agriculture and Life Sciences, Chungnam National University. The seeds were germinated and grown in plastic pots measuring 40 cm by 30 cm containing wet soil in a greenhouse maintained at a daytime temperature of 25 °C and a nighttime temperature of 18 °C. After about a week, sprouts began to appear, and when the seedlings developed approximately 3 to 4 leaves, fresh young leaves of E. indica were collected, immediately frozen in liquid nitrogen, and stored in a −80 °C deep freezer for genome sequencing. High-quality HMW DNA was extracted using the Wizard HMW DNA extraction kit. The same young leaves used for genomic DNA extraction were also utilized for DNA and RNA extraction using a Smartgene plant DNA extraction kit and a Qiagen plant mini RNA extraction kit, respectively. The quality and purity of the extracted samples were assessed using a nano-MD spectrophotometer (Scinco, Seoul, South Korea) and gel electrophoresis.
Library preparation and sequencing
The long read library was prepared using the PacBio SMARTbell prep kit 3.0 and SMARTbell barcoded adapter plate 3.0. Long-read sequencing was performed on a PacBio Revio sequencer using two Revio SMART cells, producing 18.97 Gb of raw data with an N50 length of 13.57 Kb and a total of 1,397,301 reads, covering approximately 39.7x of the genome (Table 1). The quality of the long-read sequencing data was high, with 91.48% of reads achieving a Q30 quality score. The short read library was prepared using an Illumina TruSeq DNA Nano (550 bp) kit for paired-end sequencing, and sequencing was performed on the Illumina NovaSeq 6000 platform according to the manufacturer’s instructions. This generated 170,978,570 reads, yielding raw data covering approximately 54x of the genome (Table 1). Short-read sequencing achieved a Q30 of 86.815% and an average quality score of 35.1, indicating high sequencing accuracy suitable for genome assembly.
For RNA sequencing, libraries were prepared using an Illumina TruSeq stranded mRNA kit following the manufacturer’s guidelines, and sequencing was performed on the Illumina NovaSeq 6000 platform. The RNA-seq data were of excellent quality, with 95.87% of bases achieving a Q30 score and an average quality score of 36.32, ensuring high accuracy for downstream analysis. In total, 6.84 Gb of RNA-seq data were generated (Table 1), which provided a reliable foundation for protein-coding gene prediction.
Chromosome-level genome assembly
The E. indica genome was assembled using PacBio HiFi long reads with a Phred score of Q20 or higher and NextDenovo v2.5.010. The initial draft genome consisted of 255 contigs with a total size of 513 Mb and a contig N50 of 7.33 Mb (Table 2). For chromosome-level scaffolding, a Pore-C library was prepared. This involved crosslinking with formaldehyde, nuclei isolation, chromatin digestion with NlaIII, ligation of crosslinked DNA, protein degradation, and DNA extraction using phenol:chloroform:isoamyl alcohol (25:24:1). A 2 μg DNA sample was prepared for ONT sequencing with a SQK-LSK110 ligation kit (Oxford Nanopore Technologies, UK) and sequenced on a PromethION flowcell. Guppy v6.5.711 software generated raw fastq files, which were filtered with NanoPlot v1.41.612 (Phred quality ≥ 7), resulting in 37 Gb of data with an average quality score of 15.4 and an N50 of 2.22.
The filtered Pore-C fastq files and draft assembly were processed using Pore-C Snakemake v5.5.413 to create mnd files, utilizing 11.99 Gb of data. These files, along with the initial assembly, were then input into the 3D-DNA v18092214 pipeline to produce assembly files, with specific options (e.g., -i 10000, --polisher-input-size 1000000, --splitter-input-size 1000000, -r 2, --editor-coarse-resolution 250000, --editor-coarse-region 1250000, -q 0, --polisher-coarse-resolution 1000000, --polisher-coarse-region 30000000) used to optimize the process. Manual curation in JuiceBox v1.11.0815 generated the review.assembly file, which, together with the initial files, was further processed with 3D-DNA v190716 (option -i 15000) to finalize the corrected assembly. The resulting scaffolds were anchored to nine pseudochromosomes, yielding a chromosome-scale assembly with a total length of 505 Mb (Fig. 1).

Pore-C interaction heatmap of E. indica genome. Pore-C interaction matrix showing the pairwise correlations among nine pseudochromosomes.
Genome size estimation
Before estimating the genome size, Illumina short reads were processed using Trimmomatic V0.4016 to remove low-quality reads and adapters. The genome characteristics of E. indica were assessed using a K-mer based method17. The distribution of the K-mer read depth was calculated using Jellyfish v2.3.118, extracting standard K-mers at k = 21. Genome size and heterozygosity were estimated with GenomeScope v2.017 using default parameters. The genome size of E. indica was estimated to be 478 Mb, with a heterozygosity rate of 0.68% (Fig. 2).

K-mer profile (k = 21) spectral analysis to estimate genome size.
Repeat annotation
To identify repetitive sequences, we first constructed a new repeat sequence library for the E. indica genome using RepeatModeler v2.0.419, which integrates RECON20 and RepeatScout21. We then used RepeatMasker v4.1.5 (http://www.repeatmasker.org) to search for repeats through de novo repeat libraries and homology-based repeat searches with RepBase22. LTR_FINDER v1.223 and GenomeTools v1.6.224’s LTR_harvest25 were used to identify long terminal repeat retrotransposons (LTR-RTs). LTR_retriever v2.9.026 was employed to identify intact LTR-RTs among the candidate LTR-RTs, which were then used to calculate insertion ages. Repeats accounted for 59.76% of the genome, with most repeats being class I retrotransposons. LTR elements constituted 39.93% of the genome, with Gypsy elements making up 23.76% and Copia elements 12.64%. Class II DNA transposons accounted for 2.40% of the genome (Table 3 / Fig. 3).

Genomic features of E. indica. From the outermost to innermost track, the circular plot shows chromosome scale, gene density, repeat ratio, GC content, Copia abundance, Gypsy abundance, and LTR abundance.
Gene prediction and functional annotation
The prediction of protein-coding genes in the assembled genome was performed using a combination of ab initio, homology-based, and transcriptome-based prediction methods. RNA-seq raw data were trimmed for quality using Trimmomatic v0.4016 and high-quality reads were aligned to the assembly using Hisat2 v2.2.127. Ab initio predictions were carried out using BRAKER v3.0.728 and SNAP v2006-07-2829. Protein sequences from Sorghum bicolor, Zea mays, Brachypodium distachyon, and Oryza sativa, downloaded from Phytozome, as well as Cynodon transvaalensis data provided by Dr. Xiangfeng Wang from the National Maize Improvement Center, College of Agronomy and Biotechnology, China Agricultural University, were used for homology-based gene prediction with GeMoMa v1.930. Transcriptome-based predictions utilized Cufflinks v2.2.131 and StringTie v2.2.132. The predicted genes were integrated using EvidenceModeler (EVM) v2.0.033.
To investigate the functions of the 26,836 predicted genes, they were queried against the NCBI viridiplantae protein non-redundant (nr)34, Uniprot35, and EggNOG-mapper36, Gene Ontology (GO)37, (KEGG)38, and Pfam39 databases using DIAMOND v2.1.940. From the results, 97.01%, 96.12%, 92.24%, 37.85%, 41.99%, and 81.35% of the protein-coding genes were annotated in the nr, Uniprot, eggNOG, GO, KEGG, and Pfam databases, respectively (Table 4).
Data Records
The Illumina, PacBio, Pore-C, and RNA-Seq data of E. indica reported in this study are available in the NCBI SRA database under the project accession SRP51096341. The accession numbers for the Illumina, PacBio, Pore-C, and RNA-Seq data are SRR29243660, SRR29243661, SRR29243662, and SRR29243659, respectively. The final chromosome assembly can be found in the NCBI GeneBank database under the WGS project ID JBEWPU01 and the GeneBank accession ID GCA_040549725.141,42. The genome annotation data have been deposited in the Figshare database43.
Technical Validation
To ensure high-quality and comprehensive assembly, we validated the Korean E. indica genome using several metrics, focusing on BUSCO v5.5.044, LAI scores, and synteny analysis with the Chinese E. indica genome assembly. The genome and RNA data for the Chinese E. indica were downloaded from NCBI GenBank (accessions JARKIM000000000 and JARKIL000000000) and CoGe (accession numbers id66361 and id66364), respectively, both representing the Chinese E. indica species.
-
1.
Assembly Completeness: Using BUSCO v5.5.0 with the Embryophyta odb10 dataset, we confirmed that 96.8% of the orthologs were complete, with 2.47% missing and 0.68% fragmented, indicating robust gene coverage (Fig. 4). We also conducted further validation to strengthen gene annotation through BUSCO analysis on the predicted protein sequences, which confirmed the completeness of our annotation, with 88.8% complete BUSCOs, 87.2% single-copy BUSCOs, and 1.6% duplicated BUSCOs (Table 5). This high completeness score is further supported by RNA-Seq data analysis using HiSat245, StringTie32, and gffcompare46, which revealed an exon sensitivity of 69.3% and an intron sensitivity of 81.0%. Although transcript-level sensitivity was 33.5%, this is consistent with known challenges of capturing complex alternative splicing patterns using RNA-Seq data. This analysis identified 23.1% novel loci and 18.8% novel exons, contributing to previously unannotated gene elements (Table 6).
Fig. 4 
BUSCO analysis evaluated both genome assembly and protein-coding gene predictions, showing over 98.7% completeness, indicating high-quality results.
Table 5 Summary of BUSCO and RNA-Seq validation metrics for the Korean E. indica genome. Table 6 RNA-Seq sensitivity metrics for assembly completeness of the Korean E. indica genome. -
2.
Genome Integrity: The LAI score, calculated using LTR_retriever26, averaged 17.75, underscoring the structural robustness of the Korean assembly. This score is comparable to the Chinese assemblies, demonstrating the reliability of the assembly across genomic regions (Table 7).
Table 7 Comparison of key genome assembly metrics between Korean and Chinese E. indica assemblies. -
3.
Comparative Synteny and Annotation Quality: Synteny analysis conducted with MCScanX47 revealed that 65.63% of genes are collinear between the Korean and Chinese E. indica GS genomes (Fig. 5), confirming a high level of conservation in gene order while also emphasizing structural variations unique to the Korean population. The gene annotation comparison showed a close alignment with the Chinese assemblies in terms of gene count and scaffold N50 (58.9 Mb vs. 57 Mb). To further assess the quality of our annotation, we compared key metrics between the Korean E. indica assembly and the Chinese GS and GR genomes. Although the gene count and exon structure are broadly similar, our annotation offers new insights into the evolutionary adaptations specific to the Korean E. indica population (Table 8). Together, the synteny analysis, BUSCO validation, and RNA-Seq alignment confirm the structural integrity and completeness of our assembly, showcasing both conserved genomic features and population-specific variations.These validation steps confirm that the Korean E. indica genome assembly is of high quality and contributes valuable genetic diversity insights, laying the foundation for future pan-genome studies.
Fig. 5 
Synteny plot between Korean and glyphosate-sensitive E. indica genomes. This synteny plot compares the Korean E. indica genome (y-axis) with the glyphosate-sensitive E. indica genome assembled in 20238 (x-axis). The diagonal lines indicate regions of conserved gene order (collinearity) between the two genomes, while off-diagonal elements represent structural variations, such as inversions or translocations, reflecting genetic differences between the populations.
Table 8 Gene structure comparison between Korean and Chinese E. indica Assemblies.


Usage Notes
The chromosome-level genome assembly of the Korean E. indica population presented in this study provides a critical resource for understanding the genetic diversity and adaptive traits of this globally distributed and invasive weed species. While a high-quality E. indica genome from a Chinese population was published in 20238, our research focuses on a geographically distinct population in South Korea, known for its high genetic variability due to its weedy origin. The genetic differences between populations from distinct geographical regions are significant for several reasons:
-
1.
Ecological and Evolutionary Insights: Genetic diversity across E. indica populations can reveal how different environmental pressures, such as climate, soil composition, and agricultural practices, drive local adaptations. This understanding is essential for developing strategies to manage E. indica as a weed in various regions, particularly in agriculture-intensive areas.
-
2.
Population-Specific Adaptations: By studying a Korean population, researchers can explore genetic mechanisms specific to this region, such as resistance to local herbicides, tolerance to regional stress factors (e.g., temperature or drought), and unique reproductive strategies. These insights are crucial for developing population-specific management and control measures.
-
3.
Comparative Genomics and Pan-genome Studies: The data provided here lay the groundwork for future pan-genome projects that aim to capture the full genetic diversity of E. indica. Researchers can use this assembly in comparative studies with other E. indica genomes to investigate structural variations, gene family expansions or contractions, and evolutionary processes. This is especially relevant for understanding the genetic basis of traits like invasiveness and herbicide resistance.
Recommendations for Data Use: Researchers interested in comparative genomic analyses can integrate this assembly with the previously published Chinese genome to identify population-specific genetic features. We recommend using bioinformatics tools such as MCScanX for synteny analysis, OrthoFinder48 for orthologous gene comparisons, and CAFE49 for investigating gene family evolution. For those studying ecological adaptation or weed management strategies, the genome data can be used to identify genes linked to stress responses or metabolic pathways relevant to herbicide resistance.
Limitations and Considerations: While this assembly provides a robust and high-quality resource, users should consider that genetic variation may exist even within the Korean population. Additionally, environmental factors specific to South Korea may have shaped unique adaptations that may not be present in other regions.
Potential Applications: This genome assembly can aid in breeding programs for crop protection, the development of region-specific herbicide resistance management strategies, and evolutionary studies of the Eleusine genus. Furthermore, our dataset complements existing genomic resources, enriching the overall understanding of E. indica’s adaptability and invasiveness.
Code availability
No specific script was used in this work. All bioinformatics tools used in this study followed their respective protocols and manuals. If specific parameters are not mentioned, the default parameters were used. The versions of the software used are indicated in the Methods section.
References
Wu, H.-W., Jiang, W.-L. & Yan, M. Goosegrass (Eleusine indica) density effects on cotton (Gossypium hirsutum). Journal of integrative agriculture 14, 1778–1785 (2015).
Luchian, V., Georgescu, M. I., Săvulescu, E. & Popa, V. Some aspects of morpho-anatomical features of the invasive species Eleusine indica (L.) Gaertn. Scientific Papers. Series A. Agronomy 62 (2019).
Chen, S., McElroy, J. S., Dane, F. & Peatman, E. Optimizing transcriptome assemblies for Eleusine indica leaf and seedling by combining multiple assemblies from three de novo assemblers. The plant genome 8, plantgenome2014.2010.0064 (2015).
Alcantara, R., Fernandez, P., Smeda, R. J., Alves, P. L. & De Prado, R. Response of Eleusine indica and Paspalum distichum to glyphosate following repeated use in citrus groves. Crop Protection 79, 1–7 (2016).
Loddo, D. et al. First report of glyphosate-resistant biotype of Eleusine Indica (L.) Gaertn. in Europe. Agronomy 10, 1692 (2020).
Plaza, G., Hoyos, V. & Vázquez-García, J. G. Alcántara-de la Cruz, R. & De Prado, R. First case of multiple resistance to EPSPS and PSI in Eleusine indica (L.) Gaertn. collected in rice and herbicide-resistant crops in Colombia. Agronomy 11, 96 (2021).
Deng, W. et al. Cyhalofop-butyl and glyphosate multiple-herbicide resistance evolved in an Eleusine indica population collected in Chinese direct-seeding rice. Journal of agricultural and food chemistry 68, 2623–2630 (2020).
Zhang, C. et al. Subtelomeric 5-enolpyruvylshikimate-3-phosphate synthase copy number variation confers glyphosate resistance in Eleusine indica. Nature Communications 14, 4865, https://doi.org/10.1038/s41467-023-40407-6 (2023).
Kerr, R. A. Goosegrass biology, genetic diversity and innovative control measures, Clemson University, (2019).
Hu, J. et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biology 25, 107 (2024).
Wick, R. R., Judd, L. M. & Holt, K. E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome biology 20, 1–10 (2019).
De Coster, W. & Rademakers, R. NanoPack2: population-scale evaluation of long-read sequencing data. Bioinformatics 39, https://doi.org/10.1093/bioinformatics/btad311 (2023).
Ulahannan, N. et al. Nanopore sequencing of DNA concatemers reveals higher-order features of chromatin structure. bioRxiv, 833590 https://doi.org/10.1101/833590 (2019).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Systems 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120, https://doi.org/10.1093/bioinformatics/btu170 (2014).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature communications 11, 1432 (2020).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome research 12, 1269–1276 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358, https://doi.org/10.1093/bioinformatics/bti1018 (2005).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic acids research 35, W265–W268 (2007).
Gremme, G., Steinbiss, S. & Kurtz, S. GenomeTools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM transactions on computational biology and bioinformatics 10, 645–656 (2013).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18, https://doi.org/10.1186/1471-2105-9-18 (2008).
Ou, S. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol 176, 1410–1422, https://doi.org/10.1104/pp.17.01310 (2018).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Bruna, T., Hoff, K., Lomsadze, A., Stanke, M. & Borodovsky, M. (2021).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59, https://doi.org/10.1186/1471-2105-5-59 (2004).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods Mol Biol 1962, 161–177, https://doi.org/10.1007/978-1-4939-9173-0_9 (2019).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols 7, 562–578, https://doi.org/10.1038/nprot.2012.016 (2012).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Sayers, E. W. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res 50, D20–d26, https://doi.org/10.1093/nar/gkab1112 (2022).
Consortium, T. U. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Research 49, D480–D489, https://doi.org/10.1093/nar/gkaa1100 (2020).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol Biol Evol 38, 5825–5829, https://doi.org/10.1093/molbev/msab293 (2021).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nature Genetics 25, 25–29, https://doi.org/10.1038/75556 (2000).
Kotera, M., Hirakawa, M., Tokimatsu, T., Goto, S. & Kanehisa, M. The KEGG databases and tools facilitating omics analysis: latest developments involving human diseases and pharmaceuticals. Methods Mol Biol 802, 19–39, https://doi.org/10.1007/978-1-61779-400-1_2 (2012).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res 49, D412–d419, https://doi.org/10.1093/nar/gkaa913 (2021).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12, 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP510963 (2024).
NCBI GeneBank https://identifiers.org/ncbi/insdc.gca:GCA_040549725.1 (2024).
Lee, S. E. indica Annotation https://doi.org/10.6084/m9.figshare.25940917.v1 (2024).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molecular Biology and Evolution 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Pertea, G. & Pertea, M. GFF utilities: GffRead and GffCompare. F1000Research 9 (2020).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic acids research 40, e49–e49 (2012).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biology 20, 238, https://doi.org/10.1186/s13059-019-1832-y (2019).
De Bie, T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271, https://doi.org/10.1093/bioinformatics/btl097 (2006).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT, No. 2022R1A2C1004127).
Author information
Authors and Affiliations
Contributions
C.K. conceptualized and designed the study. S.L. prepared the plant samples, conducted the experiments, performed data analysis, and drafted the manuscript. All authors read, edited, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, S., Kim, C. Chromosome-scale genome assembly of Korean goosegrass (Eleusine indica). Sci Data 12, 156 (2025). https://doi.org/10.1038/s41597-025-04490-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04490-2
