
Abstract
Raman spectra play an important role in characterizing two-dimensional materials, as they provide a direct link between the atomic structure and the spectral features. In this work, we present an automatic computational workflow for Raman spectra using all-electron density functional perturbation theory. Utilizing this workflow, we have successfully completed the Raman spectra calculation for 3504 different two-dimensional materials, with the resultant data saved in a data repository.
Similar content being viewed by others

Background & Summary
Atomically thin two-dimensional (2D) materials are regarded as promising candidates for various applications, including electronics, optoelectronics, sensors and flexible devices. This is attributed to their unique characteristics, which include high carrier mobility, tunable bandgaps, large specific surface area, and thickness at the atomic level1,2,3. To date, more than sixty compounds have been synthesized or exfoliated as monolayers, including well-known semiconductors (e.g. metallic transition metal dichalcogenides (TMDCs), phosphorene)4,5, insulators (e.g. GaN, BN)6,7, semi-metals and metals (e.g. graphene, silicene, borophene)8,9. However, the already known monolayer 2D materials are only the tip of a much larger iceberg. In fact, recent data mining studies predict that more than 6,000 monolayer 2D materials could be exfoliated from known layered bulk crystals or synthesized experimentally directly by chemical vapor deposition and molecular beam epitaxy10,11,12.
To accelerate discovery and deployment of these novel advanced 2D materials and realize the increasing requirements for these functional applications, materials preparation and structures characterization are the first prerequisite. Raman spectroscopy is a versatile technique for probing the vibrational modes of molecules and crystals from inelastically scattered light and is widely used for identifying materials through their unique vibrational fingerprints13. As a high-sensitivity and nondestructive spectral analysis method, Raman spectra can quickly characterize a lot of features, such as chemical composition, layer thickness, interlayer coupling, strain, crystal symmetries, and sample quality. It has taken a long time to develop the theoretical methods and numerical algorithms to calculate Raman spectroscopy. The ab-initio computational scheme based on full quantum mechanical treatment has been successfully deployed to calculate Raman spectra of both molecules and solids14,15,16,17,18. As a quantitatively accurate description, its calculated Raman spectra show a good agreement with the experimentally measured spectra13,17,18,19,20. In comparison with the experimental measurement of Raman spectroscopy, the high-throughput computation of Raman spectra will be more cost-effective and convenient. Systematic calculations will lead to a standard data set, facilitating the identification and classification of material structures, the discovery of correlations, and the application of machine learning techniques to identify deep and complex correlations in the materials space. Therefore, the compilation of a comprehensive library of Raman spectra of 2D materials across different crystal structures and chemical compositions is a critical and timely endeavor.
In this work, we design an efficient ab initio high-throughput workflow to calculate Raman spectra of monolayer 2D materials, which allow us to calculate structures with more than thirty atoms. Our calculation method is based on all-electron density functional perturbation theory (DFPT) implementation in FHI-aims software employing a localized atomic orbital (LCAO) basis set. The initial monolayer structures for two-dimensional materials are from 2D Materials Encyclopedia (2DMatPedia) database (http://www.2dmatpedia.org/.)10. Their structural, electronic and energetic properties have been obtained by high-throughput calculations. We focus on the first-order Raman processes in which only a single phonon is involved. Typically, this is the dominant scattering process in perfect crystals. We use the Tianhe supercomputer to calculate the harmonic Raman spectra of 3504 two-dimensional materials. Our work is not only a valuable reference for experimentalists and theoreticians working in the field of 2D materials, but also represents a step in the direction of autonomous (in situ) characterization of materials.
Methods
Computational workflow
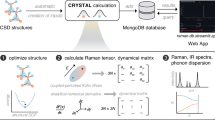
We use the DFPT implementation in the FHI-aims software package18,21 to evaluate the polarizability, as illustrated in Fig. 1(a). For more information, please refer to Supplementary information (S1). After completing the ground state DFT calculation, the ground state density of the system n(0)(r) is obtained. The DFPT cycle then begins by using an initial guess for the response of the density matrix P(1), which allows the construction of the respective density n(1)(r). The associated response of the electrostatic potential \({V}_{es,tot}^{(1)}({\bf{r}})\) is calculated by solving the Poisson equation in real space. The response Hamiltonian H(1) is then calculated using the response density and potential. With these ingredients, the Sternheimer equation can be set up. Solving the Sternheimer equation allows the update of the response expansion coefficients C(1). To accelerate convergence, a linear or more efficient Pulay-mixing scheme22 is employed. The DFPT loop is iteratively repeated until self-consistency is reached, i.e., until the changes in P(1) become smaller than a user-given threshold. Finally, physical properties such as the polarizability tensor α are evaluated using the converged response density matrix. Our Raman spectrum calculation workflow is shown in Fig. 1(b). The data is obtained from the 2DMatPedia database10 and using the DFPT method21 to calculate the Raman spectrum. The overall calculation is divided into four steps: first, we obtain the material structure data in POSCAR format from the 2DMatPedia database10, and convert the data format to the format that FHI-aims can read. In the second step, we optimized all the material structures. Since the calculation time is proportional to the number of atoms in the structure, we split the structure of the materials that contain more than 15 atoms. The split structures are divided into multiple computation nodes for parallel calculation, thus reducing the calculation time. Third, FHI-aims is used to calculate the harmonic Raman spectrum of the finite difference method. Finally, we summarize the data and upload it to the cloud database for users to access. The above calculation process is automated and can be executed in parallel on a large scale.

Computational workflow of the harmonic Raman spectra based on all-electron DFPT calculation in FHI-aims.
The k-point density was determined by dividing 50 Å by the lattice constant, and a vacuum spacing of approximately 20 Å was introduced to minimize interactions between periodic images. We employed the Tkatchenko-Scheffler method with iterative Hirshfeld partitioning to calculate the vdW corrections23. The vdW correction is mainly used for interlayer interactions. For monolayers, increasing the vdW correction usually does not affect the calculations, but in some complex 2D systems, dispersion forces cannot be ignored. Increasing the vdW correction can ensure that these systems are calculated correctly. A scalar relativistic treatment with the atomic zeroth order regular approximation(ZORA) was included in calculations24. All of our calculations were performed with ‘light’ numerical settings and basis-set configurations within the FHI-aims code. In the structure optimization, the convergence tolerance of energy, charge density, and sum of eigenvalues were set to 1 × 10−6 eV, 1 × 10−6e/Å3 and 1 × 10−4 eV. The structures were relaxed with PBE functional plus vdw correction using a Broyden-Fletcher-Goldfarb-Shanno (BFGS) optimization algorithm until the maximum force on each atom was less than 0.01 eV/ Å3. To ensure the accuracy of our calculations, we optimized all the 2D structures by introducing a vacuum layer, following the approach used in the Materials Encyclopedia (2DMatPedia) database.
We calculated harmonic Raman simulation through the finite differences method and obtain the polarizability tensor from the DFPT theory18. We calculate the polarizability tensors with the local density approximation (LDA) functional given that we have previously shown in this article18 that it is much cheaper and this method will not cause obvious accuracy loss of the Raman spectrum. The convergence tolerance of energy, charge density, sum of eigenvalues were set to 1 × 10−8 eV, 1 × 10−8e/Å3, 1 × 10−6 eV.
Data Records
Computational data record
The final database contains geometry data, Raman frequencies, and intensities. These are stored in separate files and can be downloaded directly from the Materials Cloud Archive25. The data can also be browsed online in Computational Raman Database website (http://mathtc.nscc-tj.cn/raman).
Technical Validation
We validated our computational results by comparing them with experimental Raman spectra of 12 different structures13. The 12 materials are BN19, Graphene26, MoS227, MoTe228, MoTe2T29, Pd2Se430, Phosporene31, PtS232, PtSe233, WS234, WSe227, WTe235. The comparison results are shown in Fig. 2. The discrepancies between our calculated results and experimental measurements can be attributed to limitations in our calculation method. In our current approach, we only considered harmonic vibrations and did not account for non-harmonic effects, substrate influences, or excitonic effects. To address these shortcomings, we plan to incorporate non-harmonic effects and perform high-precision calculations in future iterations of the database. Furthermore, we recognize that experimental measurement conditions also contribute to the discrepancies between our theoretical predictions and experimental observations. Experimental measurements often employ angle-resolved Raman spectroscopy, which differs from the theoretical principles used in our calculations. The constraints imposed by experimental conditions, such as specific scattering angles and excitation frequencies, may result in certain Raman peaks being undetected. We have made a general analysis for the whole database, which is added into Supplementary Information (S2), and we derived the physical descriptor of the Raman shift for the binary materials. Our Raman spectrum simulation provides the orientation-averaged “powder" Raman spectrum. Our program can also calculate the angle-resolved polarized Raman spectra by exporting the Raman tensor, although this aspect has not been the primary focus of our current work. We have added the discussion about the angle-resolved polarized Raman spectra in the Supplementary Information (S3). We have also compared our results with those presented in Ref. 13. The primary methodological difference lies in the calculation approach: we use Density Functional Perturbation Theory (DFPT), where Raman tensors are computed via mixed second derivatives of the total energy with respect to atomic displacement and electric field. In contrast, Ref. 13 employs time-dependent perturbation theory, deriving Raman intensities from transition matrix elements that involve electron-phonon coupling terms, summed over intermediate states. These two approaches use different finite difference steps, which may result in variations in the calculated vibrational frequencies. Furthermore, our work utilizes all-electron full-potential calculations for both geometry optimization and Raman calculations, while Ref. 13 employs the PAW (Projector Augmented Wave) method, which can lead to slight structural differences affecting the computed frequencies. For direct comparison, we have included the results from Ref. 13 (represented by gray lines) in Fig. 2.

The comparison of experimental and computational Raman spectra of 12 different materials. The blue solid line shows our computational results, while the gray solid line shows the theoretical calculations from Taghizadeh et al.13, and the red dashed line represents the experimental data.
Usage Notes
Two dimensional materials have many unique properties compared with other materials. We can quickly determine the material composition by comparing the experimental Raman spectrum data with our calculated Raman spectrum data set. At the same time, our data set also provides basic data for reducing the cost of Raman spectroscopy calculation through machine learning. At the same time, we will open source the automated computing workflow program used in the computing process. These programs are not limited to the calculation of two-dimensional material Raman spectra, and can help other researchers with high-throughput computing needs to reduce their workload. All calculated Raman spectra are open access and sharing in the our built website (http://mathtc.nscc-tj.cn/raman/) and also in our Materials Cloud Archive database25.
Code availability
The FHI-aims code is used for DFT calculation and DFPT simulation calculation. The workflow used in Fig. 2 is completed by using the python scripts, which can execute, manage and record the workflow. The link for these tools is: https://github.com/DownFly/FHI-aims-Raman-calculation-Script.
References
Tang, L., Tan, J., Nong, H., Liu, B. & Cheng, H.-M. Chemical vapor deposition growth of two-dimensional compound materials: controllability, material quality, and growth mechanism. Accounts of Materials Research 2, 36–47 (2020).
Mounet, N. et al. Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds. Nature nanotechnology 13, 246–252 (2018).
Zhang, L., Dong, J. & Ding, F. Strategies, Status, and Challenges in Wafer Scale Single Crystalline Two-Dimensional Materials Synthesis. Chemical reviews 121, 6321–6372 (2021).
Zhou, J. et al. A library of atomically thin metal chalcogenides. Nature 556, 355–359 (2018).
Zhou, J. et al. Composition and phase engineering of metal chalcogenides and phosphorous chalcogenides. Nature Materials 22, 450–458 (2023).
Al Balushi, Z. Y. et al. Two-dimensional gallium nitride realized via graphene encapsulation. Nature Materials 15, 1166–1171 (2016).
Xu, R., Zou, X., Liu, B. & Cheng, H.-M. Computational design and property predictions for two-dimensional nanostructures. Materials Today 21, 391–418 (2018).
Zhang, S. et al. Recent progress in 2D group-VA semiconductors: from theory to experiment. Chemical Society Reviews 47, 982–1021 (2018).
Wu, R. et al. Micrometre-scale single-crystalline borophene on a square-lattice Cu (100) surface. Nature Chemistry 14, 377–383 (2022).
Zhou, J. et al. 2DMatPedia, An open computational database of two-dimensional materials from top-down and bottom-up approaches. (2019).
Haastrup, S. et al. The Computational 2D Materials Database: high-throughput modeling and discovery of atomically thin crystals. 2D Materials 5, 042002 (2018).
Zavabeti, A. et al. A liquid metal reaction environment for the room-temperature synthesis of atomically thin metal oxides. Science 358, 332–335 (2017).
Taghizadeh, A., Leffers, U., Pedersen, T. G. & Thygesen, K. S. A library of ab initio Raman spectra for automated identification of 2D materials. Nature Communications 11, 3011 (2020).
Liang, Q., Dwaraknath, S. & Persson, K. A. High-throughput computation and evaluation of raman spectra. Scientific Data 6, 1–7 (2019).
Neugebauer, J., Reiher, M., Kind, C. & Hess, B. A. Quantum chemical calculation of vibrational spectra of large molecules-Raman and IR spectra for buckminsterfullerene. Journal of Computational Chemistry 23, 895–910 (2002).
Veithen, M., Gonze, X. & Ghosez, P. Nonlinear optical susceptibilities, Raman efficiencies, and electro-optic tensors from first-principles density functional perturbation theory. Physical Review B 71, 125107 (2005).
Maschio, L., Kirtman, B., Rérat, M., Orlando, R. & Dovesi, R. Ab initio analytical Raman intensities for periodic systems through a coupled perturbed Hartree-Fock/Kohn-Sham method in an atomic orbital basis. II. Validation and comparison with experiments. The Journal of chemical physics 139, 164102 (2013).
Shang, H. et al. All-electron, real-space perturbation theory for homogeneous electric fields: theory, implementation, and application within DFT. New Journal of Physics 20, 073040 (2018).
Cai, Q. et al. Raman signature and phonon dispersion of atomically thin boron nitride. Nanoscale 9, 3059–3067 (2017).
Neumann, C. et al. Raman spectroscopy as probe of nanometre-scale strain variations in graphene. Nature Communications 6, 8429 (2015).
Shang, H., Carbogno, C., Rinke, P. & Scheffler, M. Lattice dynamics calculations based on density-functional perturbation theory in real space. Computer Physics Communications 215, 26–46 (2017).
Pulay, P. Convergence acceleration of iterative sequences. The case of SCF iteration. Chemical Physics Letters 73, 393–398 (1980).
Tkatchenko, A. & Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Physical Review Letters 102, 073005 (2009).
Blum, V. et al. Ab initio molecular simulations with numeric atom-centered orbitals. Computer Physics Communications 180, 2175–2196 (2009).
Li, G. et al. High-throughput computation of ab initio Raman spectra for two-dimensional materials (2023).
Wu, J.-B., Lin, M.-L., Cong, X., Liu, H.-N. & Tan, P.-H. Raman spectroscopy of graphene-based materials and its applications in related devices. Chemical Society Reviews 47, 1822–1873 (2018).
Yang, M. et al. Anharmonicity of monolayer MoS2, MoSe2, and WSe2: A Raman study under high pressure and elevated temperature. Applied Physics Letters 110, 093108 (2017).
Ruppert, C., Aslan, B. & Heinz, T. F. Optical Properties and Band Gap of Single- and Few-Layer MoTe2 Crystals. Nano letters 14, 6231–6236 (2014).
Gupta, U. et al. Characterization of few-layer 1T-MoSe2 and its superior performance in the visible-light induced hydrogen evolution reaction. APL Materials 2, 092802 (2014).
Yu, J. et al. Direct Observation of the Linear Dichroism Transition in Two-Dimensional Palladium Diselenide. Nano letters 20, 1172–1182 (2020).
Xia, F., Wang, H. & Jia, Y. Rediscovering black phosphorus as an anisotropic layered material for optoelectronics and electronics. Nature Communications 5, 4458 (2014).
Zhao, Y. et al. Extraordinarily Strong Interlayer Interaction in 2D Layered PtS2. Advanced materials 28, 2399–2407 (2016).
Yan, M. et al. High quality atomically thin PtSe2 films grown by molecular beam epitaxy. 2D Materials 4, 045015 (2017).
Liu, H.-L. et al. Deep-ultraviolet Raman scattering spectroscopy of monolayer WS2. Scientific reports 8, 11398 (2018).
Chen, K. et al. A Simple Method for Synthesis of High-Quality Millimeter-Scale 1T’ Transition-Metal Telluride and Near-Field Nanooptical Properties. Advanced Materials 29, 1700704 (2017).
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. T2222026), and was supported as part of A Multi-Scale and High-Efficiency Computing Platform for Advanced Functional Materials, a scientific research program funded by Haihe Laboratory in Tianjin, Grants No. 22HHXCJC00007. The authors acknowledge National Supercomputer Center in Tianjin, and the calculations were performed on Tianhe 3F. This work is funded by the opening fund of Key Laboratory of Rare Earths, Chinese Academy of Sciences.
Author information
Authors and Affiliations
Contributions
G. Li, Y.X. Gao, D.Y. Xie and L.L. Zhu performed the Raman spectra calculations, developed the code for visualization, performed the data benchmarking and analysis, and wrote the paper. H.H. Shang contributed to supervision of the calculations, the data analysis and take the lead in writing the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, G., Gao, Y., Xie, D. et al. High-Throughput Computation of ab initio Raman Spectra for Two-Dimensional Materials. Sci Data 12, 373 (2025). https://doi.org/10.1038/s41597-025-04593-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04593-w
This article is cited by
-
IR-NMR multimodal computational spectra dataset for 177K patent-extracted organic molecules
Scientific Data (2025)
