
Abstract
Coral reefs are among the most biodiverse and economically significant ecosystems globally, yet they are increasingly degrading due to global climate change and local human activities. The sun coral Tubastraea coccinea (T. coccinea) an obligate heterotroph lacking symbiotic zooxanthellae, exhibits remarkable tolerance to conditions that cause bleaching and mortality in zooxanthellate species. With its extensive low-latitude distribution across multiple oceans, T. coccinea has become a highly invasive species, adversely impacting native species, degrading local ecosystems, and causing significant socio-economic challenges that demand effective management. Despite substantial research efforts, the molecular biology of T. coccinea remains insufficiently characterized. To address this gap, we generated a draft genome assembly for T. coccinea using PacBio Hi-Fi long-read sequencing. The assembly spans 875.9 Mb with a scaffold N50 of 694.3 kb and demonstrates high completeness, with a BUSCO score of 97.4%. A total of 37,307 protein-coding sequences were identified, 95.2% of which were functionally annotated through comparisons with established protein databases. This reference genome provides a valuable resource for understanding the genetic structure of T. coccinea, advancing research into its adaptive mechanism to environmental changes, and informing conservation and management strategies to mitigate its invasive impact.
Similar content being viewed by others

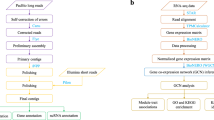

Background & Summary
Coral reefs are widely regarded as one of the most biologically diverse and ecologically fragile ecosystems on Earth1. These vital habitats support at least 25% of the world’s marine species, despite covering less than 0.2% of the ocean floor2,3. In addition to their immense ecological importance, coral reefs sustain the livelihoods of millions of people through industries such as fishing and tourism4. However, the inherent fragility of coral reefs makes them particularly susceptible to a range of anthropogenic and environmental stressors. Factors such as rising sea temperatures, ocean acidification, pollution, and destructive fishing practices pose significant threats to their survival5,6,7,8. Notably, the frequency of coral bleaching events, a major indicator of reef health, has increased globally9,10 and is expected to intensify in the coming decades11,12.
Meanwhile, the globalization of trade, tourism, and economies has exacerbated the spread of invasive species, which pose a substantial threat to biodiversity by disrupting ecosystem functions and altering community composition. Such invasions can lead to severe economic consequences13,14. One particularly concerning invasive species is Tubastraea coccinea (T. coccinea), an azooxanthellate coral species (Fig. 1), exhibits a widespread, low-latitude distribution across multiple ocean basins due to its tolerance of conditions that cause bleaching and mortality in zooxanthellate corals15. Native to the Indo-Pacific region, T. coccinea has successfully invaded various areas of the eastern Pacific, as well as the western and eastern Atlantic, extending to southern Brazil, resulting considerable environmental, economic, and social impacts16. Its highly invasive nature is facilitated by a suite of biological traits, including rapid growth, early reproductive maturity, multiple reproductive strategies, and the absence of natural predators. As a result, T. coccinea has colonized over 95% of available surfaces in the Atlantic Ocean16,17,18. Without the development of effective control measures, its spread is likely to continue unabated.

An image of the T. coccinea sample utilized for genome sequencing.
The phylogenetics of scleractinian corals remains a complex and poorly understood area of research. Despite the use of classical morphological classifications and molecular phylogenetic techniques, many aspects of coral evolution are still shrouded in uncertainty19,20,21. The ancestral state of scleractinians—whether they were originally photosymbiotic or azooxanthellate—remains controversial22,23,24,25. In part, previous studies have tended to focus on shallow-water, photosymbiotic species, and as a result, the biological diversity and ecological significance of azooxanthellate corals—comprising approximately half (>700) of all scleractinian species—remain underexplored. These corals exhibit broad distributions and notable biological diversity26,27, highlighting the need for more genetic data on this poorly understood group.
Genomic approaches have emerged as a powerful tool for advancing our understanding of coral phylogenetics and informing conservation strategies for non-model organisms28,29,30. To better understand the genetic basis of environmental adaptation and the extreme invasiveness of this particular coral genus, we present the first draft genome assembly of T. coccinea generated using long-read PacBio HiFi sequencing. The genome size of T. coccinea is 875.9 Mb, consisting of 2,573 scaffolds with an N50 length of 694.3 kb. Repetitive sequences constitute 26.01% of the total assembly, with unclassified repeats (8.75%), DNA elements (7.11%), and long interspersed nuclear elements (3.83%) (Table 1). We identified 37,307 protein-coding sequences, of which 35,221 (95.2%) are functionally annotated using five functional databases (SwissProt, KEGG, NR, GO, Pfam). The completeness of the genome, assessed using the BUSCO tool, was 96.9%, with 94.7% of the genes being complete, 2.7% fragmented, and 2.6% missing. Additionally, we predicted 1, 963 non-coding RNAs (58 miRNAs, 14,111 tRNAs, 923 rRNAs, and 224 snRNAs) in the T. coccinea genome assembly. These genomic resources will serve as a foundation for future research on the genetic mechanisms underlying the adaptability of T. coccinea to varying environmental conditions, as well as its invasive behavioral and ecological impacts.
Methods
Sample collection and DNA extraction
Tubastraea coccinea (Fig. 1) specimens were purchased from commercial suppliers in Qingdao, China, with the original source being Vietnam, and were cultured in an aquarium utilizing circulating seawater. The corals were acclimatized under laboratory conditions for 5 days prior to DNA extraction. A live specimen was further cut into 1 mm pieces and washed three times with the calcium- and magnesium-free PBS solution (wash buffer) adjusted to an osmolarity of 1,100 mOsmol. The pieces were treated with collagenase (type II, 2 mg/ml) for 30 min at room temperature to prepare cell suspensions. The cell suspension was concentrated by centrifugation (500 × g for 5 min at 4 °C). The solution was resuspended and washed three times in wash buffer. The final cell pellet was immediately fixed in liquid nitrogen for DNA extraction. Total DNA was extracted using the standard phenol/chloroform method31. The quantity of genomic DNA quantity was measured using a Nanodrop 2000 spectrophotometer, with acceptable quality standards of OD260/280 ranging from 1.8 to 2.0 and OD260/230 ranging from 2.0 to 2.2. The purity and integrity of the DNA were further assessed via 1% agarose gel electrophoresis.
Library preparation and sequencing
Qualified DNA sample were sent to Novogene (Beijing, China) for library preparation and whole genome sequencing. Using standard PacBio protocols, HiFi sequencing library was prepared with the SMRTbell™ Express Template Prep Kit 2.0 (Pacific Biosciences, California, USA) and sequencing was conducted on the Pacific Biosciences Sequel II systems (Pacific Biosciences, California, USA). The raw base-called data was transferred from the sequencer to SMRTLink v13.1 (https://www.pacb.com/support/software-downloads/), where HiFi reads were generated using the CCS algorithm. A total of 16.1 Gb of high-quality PacBio HiFi reads were obtained.
Genome assembly
The PacBio HiFi long reads were used to assemble into contigs by Hifiasm v0.16.1-r37532 with default parameters. HiFi long reads served as the input for Hifiasm to generate the primary contigs. Hifiasm attempts to eliminate haploid duplications, followed by three iterations of error correction. The assembly was examined for non-target DNA detection using Blobtools v1.1.133, where the top hit based on diamond v2.1.834 results were aligned against the NCBI nr database with an e-value cutoff of 1e-5. 69.83% of contigs showed BLAST hits to Cnidaria, while 13.92% remained unassigned and 12.90% matched to other phyla, likely due to incompleteness in the available coral genome database (Fig. 2). Genome assembly statistics was analyzed with QUAST v5.2.035 and the completeness of the genome assembly was evaluated with BUSCO v5.2.236 utilizing the conserved metazoan gene set “metazoa_odb10”. The T. coccinea assembly consisted of 875.9 Mb, across 2,573 scaffolds, with an N50 of approximately 694.3 kb and BUSCO completeness of 97.4% (Complete + Fragmented) (Fig. 3).

BlobPlot of the T. coccinea purged genome assembly. Blue dots show contigs with best blast hits to Cnidaria. Other different colors of the dots represent taxonomic information, as detailed in the legend. Histograms above and to the right of the scatter plot depict the distribution of coverage and GC content proportion, respectively.

Snail visualization summary of T. coccinea genome assembly statistics. To summarize and visualize statistics, we employed the software ‘assembly-stats’ (https://github.com/hanwnetao/snailplot-assembly-stats).
Repeat annotation
The annotation of transposable elements (TEs) and repeat sequences was conducted in two steps. Firstly, three de novo repeat identification algorithms: RepeatModeler v237 LTR_retriever v2.538, and RepeatScout v1.0.539, were applied to the T. coccinea genome assembly to build de novo repeat libraries, along with the downloading of the Repbase database40. Secondly, RepeatMasker v4.0.941 was employed to analyze and annotate the TEs and repeat sequences found in the library and the database. Software LTR_Finder v1.242 was utilized to predict long terminal repeat (LTR) sequences, with parameters ‘-D 15000 -d 1000 -L 7000 -l 100 -p 20 -C -M 0.9’, followed by LTR_retriever v2.538 to eliminate redundancy in the predicted sequences to produce nonredundant LTR sequences with default parameters.
Gene prediction and functional annotation
To achieve comprehensive gene annotation, three strategies were used for the prediction of protein-coding genes, integrating various sources of evidence: ab initio prediction, homology-based method, and transcriptome-assisted technique. For ab initio prediction, we aligned RNA-seq dataset (SRR8386108) to the T. coccinea draft genome using the STAR v2.7.143 aligner with default settings. The mapping results were subsequently utilized to generate transcript models through a combined approach involving BRAKER v2.1.544, Semi-HMM-based Nucleic Acid Parser (SNAP, v2013.11.29)45 and StringTie v2.1.646 with parameters: ‘-m 200 -a 10 -conservative -g 50 -u’. For homology-based method, metazoan protein sequences from the OrthoDB database and protein-coding sequences of several corals from The NCBI Reference Sequence Database (NCBI RefSeq), including Acropora muricata (RefSeq accession: GCF_036669905.1), Montipora foliosa (RefSeq accession: GCF_036669935.1), Pocillopora verrucose (RefSeq accession: GCF_003704095.1) and Stylophora pistillata (RefSeq accession: GCF_002571385.2), were aligned to the genome assembly utilizing TBLASTN v2.12.047 and GeneWise v2.2.048. For transcriptome-assisted technique, the RNA-seq reads were both de novo and genome-guided assembled using Trinity v2.5.149 with default parameters. The resulting transcripts were further assembled using the Program to Assemble Spliced Alignment (PASA) v2.5.250 with BLAT v3551 and GMAP v2023-12-0152 employed as aligners. Finally, the outcomes from these three strategies were integrated into a unified gene annotation using the EVidenceModeler v1.1.153. Overall, a total of 37,307 protein-coding genes were identified in the T. coccinea genome.
Utilizing the structural characteristics of tRNA, we performed de novo predictions of tRNAs using the tRNAscan-SE v2.0 software54. Additionally, rRNA, snRNA, and miRNA predictions were conducted with Infernal v1.0 software55. This analysis identified four types of noncoding RNAs: 14, 111 tRNAs, 923 rRNAs,224 snRNAs, and 58 miRNAs (Table 2).
Protein function predictions were performed using various databases, including CDD56, PANTHER57, Superfamily58, Gene3D59, SMART60, and ProSiteProfiles61 to predict protein functions by analyzing the conserved protein domains through InterProScan v5.3662. Furthermore, eggNOG-mapper v263 was utilized to search for homologous genes in the eggNOG database, enabling KEGG64 and GO65 annotation. Functional annotation of the predicted protein-coding genes was performed using blastp v2.2.26 against the SwissProt database, diamond v2.1.8 against the NR database, and hmmscan v3.3.2 against the Pfam database, with an e-value threshold of 1e-5. Ultimately, more than 35,221 (95.2%) genes were successfully annotated (Table 3).
Data Records
The raw sequencing data and genome assembly of Tubastraea coccinea have been deposited in the National Center for Biotechnology Information (NCBI) under the accession number SRR3164537766 (PacBio data) and JBJUWB00000000067 (genome assembly). Additionally, the genome annotation files (GFF and GTF), predicted protein and CDS files, as well as the gene model annotation file, are available in the figshare database68.
Technical Validation
After completing the genome assembly, we evaluated its quality based on several key aspects. (i) The assembled genome is 875.9 Mb in length, which is consistent with the previously published version and indicates a relatively complete genome. (ii) Genome coverage analysis using SAMtools v1.14 revealed 100% genome coverage and a 99.67% mapping rate for PacBio HiFi reads. (iii) The contig N50 reached 694.3 kb, which is ten times greater than that of the previous version and substantially higher than the N50 of closely related species (T. tagusensis and Tubastraea sp.), which range from 82.7 kb to 227.0 kb based on long-read sequencing69. (iv) The genome assembly completeness reached 97.4%, significantly surpassing the previous version and other Tubastraea species (T. tagusensis and Tubastraea sp.), whose completeness ranges from 88.1% to 91.6%69. (v) A BUSCO evaluation based on the Metazoa_odb10 dataset, which contains 954 conserved genes, showed a gene model completeness of 97.4%, with 94.7% of genes complete, 2.7% fragmented, and 2.6% missing. Together, these results confirm the T. coccinea genome assembly we obtained is of high-quality.
Code availability
All bioinformatics software employed in this research was executed following the manuals and protocols provided by the respective developers, with specific versions and parameters documented in the Methods section. In cases where parameters were not explicitly specified, default settings were applied. Furthermore, no custom code was developed or implemented in this study.
References
Graham, N. A. J. et al. Dynamic fragility of oceanic coral reef ecosystems. Proc. Natl. Acad. Sci. U.S.A. 103, 8425–8429, https://doi.org/10.1073/pnas.0600693103 (2006).
Knowlton, N. et al. Coral reef biodiversity. in Life in the World’s Oceans: Diversity, Distribution, And Abundance (ed. Mclntyre, A.) Ch. 4 https://doi.org/10.1002/9781444325508.ch4 (Wiley–Blackwell, 2010).
Connell, J. H. Diversity in tropical rain forests and coral reefs. Science 199, 1302–1310, https://doi.org/10.1126/science.199.4335.1302 (1978).
Moberg, F. & Folke, C. Ecological goods and services of coral reef ecosystems. Ecol. Econ. 29, 215–233, https://doi.org/10.1016/S0921-8009(99)00009-9 (1999).
Chen, P. Y., Chen, C. C., Chu, L. & McCarl, B. Evaluating the economic damage of climate change on global coral reefs. Glob. Environ. Change 30, 12–20, https://doi.org/10.1016/j.gloenvcha.2014.10.011 (2015).
Brodie, J. E. et al. Terrestrial pollutant runoff to the great barrier reef: an update of issues, priorities and management responses. Mar. Pollut. Bull. 65, 81–100, https://doi.org/10.1016/j.marpolbul.2011.12.012 (2012).
Pandolfi, J. M., Connolly, S. R., Marshall, D. J. & Cohen, A. L. Projecting coral reef futures under global warming and ocean acidification. Science 333, 418–422, https://doi.org/10.1126/science.1204794 (2011).
Baum, G., Januar, H. I., Ferse, S. C. & Kunzmann, A. Local and regional impacts of pollution on coral reefs along the Thousand Islands north of the megacity Jakarta, Indonesia. PLoS One 10, e0138271, https://doi.org/10.1371/journal.pone.0138271 (2015).
Hughes, T. P. et al. Global warming and recurrent mass bleaching of corals. Nature 543, 373–377, https://doi.org/10.1038/nature21707 (2017).
Nakamura, T. Mass coral bleaching event in Sekisei lagoon observed in the summer of 2016. J. Jpn. Coral Reef Soc. 19, 29–40, https://doi.org/10.3755/jcrs.19.29 (2017).
van Hooidonk, R. et al. Local–scale projections of coral reef futures and implications of the Paris Agreement. Sci. Rep. 6, 39666, https://doi.org/10.1038/srep39666 (2016).
Dixon, A. M., Forster, P. M., Heron, S. F., Stoner, A. M. K. & Beger, M. Future loss of local–scale thermal refugia in coral reef ecosystems. PloS Clim. 1, e0000004, https://doi.org/10.1371/journal.pclm.0000004 (2022).
Peller, T. & Altermatt, F. Invasive species drive cross–ecosystem effects worldwide. Nat. Ecol. Evol. 8, 1087–1097, https://doi.org/10.1038/s41559-024-02380-1 (2024).
Grosholz, E. D. Ecological and evolutionary consequences of coastal invasions. Trends Ecol. Evol. 17, 22–27, https://doi.org/10.1016/s0169-5347(01)02358-8 (2002).
Glynn, P. W. et al. Reproductive ecology of the azooxanthellate coral Tubastraea coccinea in the Equatorial Eastern Pacific: Part V. Dendrophylliidae. Mar Biol. 153, 529–544, https://doi.org/10.1007/s00227-007-0827-5 (2008).
Creed, J. C. et al. The invasion of the azooxanthellate coral Tubastraea (Scleractinia: Dendrophylliidae) throughout the world: History, pathways and vectors. Biol. Invasions 19, 283–305, https://doi.org/10.1007/s10530-016-1279-y (2017).
Bastos, N., Poubel Tunala, L. & Coutinho, R. Life history strategy of Tubastraea spp. corals in an upwelling area on the Southwest Atlantic: growth, fecundity, settlement, and recruitment. PeerJ 12, e17829, https://doi.org/10.7717/peerj.17829 (2024).
Sammarco, P. W., Porter, S. A., Genazzio, M. & Sinclair, J. Success in competition for space in two invasive coral species in the western Atlantic – Tubastraea micranthus and T. coccinea. PloS one 10, e0144581, https://doi.org/10.1371/journal.pone.0144581 (2015).
Duncan, P. M. Revision of the families and genera of the sclerodermic Zoantharia, Ed. & H., or Madreporaria (M. Rugosa excepted). Zool. J. Linn. Soc. 18, 1–204, https://doi.org/10.1111/j.1096-3642.1884.tb02013.x (1884).
Stolarski, J. & Roniewicz, E. Towards a new synthesis of evolutionary relationships and classification of Scleractinia. J. Paleontol. 75, 1090–1108, https://doi.org/10.1017/s0022336000017157 (2001).
Juszkiewicz, D. J. et al. Phylogeography of recent Plesiastrea (Scleractinia: Plesiastreidae) based on an integrated taxonomic approach. Mol. Phylogenet. Evol. 172, 107469, https://doi.org/10.1016/j.ympev.2022.107469 (2022).
McFadden, C. S. et al. Phylogenomics, origin, and diversification of Anthozoans (Phylum Cnidaria). Syst. Biol. 70, 635–647, https://doi.org/10.1093/sysbio/syaa103 (2021).
Kayal, E. et al. Phylogenomics provides a robust topology of the major cnidarian lineages and insights on the origins of key organismal traits. BMC Evol. Biol. 18, 68, https://doi.org/10.1186/s12862-018-1142-0 (2018).
Campoy, A. N. et al. The origin and correlated evolution of symbiosis and coloniality in scleractinian corals. Front. Mar. Sci. 7, 461, https://doi.org/10.3389/fmars.2020.00461 (2020).
Gault, J. A., Bentlage, B., Huang, D. & Kerr, A. M. Lineage-specific variation in the evolutionary stability of coral photosymbiosis. Sci. Adv. 7, eabh4243, https://doi.org/10.1126/sciadv.abh4243 (2021).
Cairns, S. D. Phylogenetic list of 722 valid Recent azooxanthellate scleractinian species, with their junior synonyms and depth ranges. Supplement to Cold-Water Corals: The Biology and Geology of Deep-Sea Coral Habitats. (Cambridge University Press, 2009).
Cairns, S. D. Species richness of recent Scleractinia. Atoll Res. Bull. 1, 459–46, https://doi.org/10.5479/si.00775630.459.1 (1999).
Allendorf, F. W., Hohenlohe, P. A. & Luikart, G. Genomics and the future of conservation genetics. Nat. Rev. Genet. 11, 697–709, https://doi.org/10.1038/nrg2844 (2010).
Formenti, G. et al. The era of reference genomes in conservation genomics. Trends Ecol. Evol. 37, 197–202, https://doi.org/10.1016/j.tree.2021.11.008 (2022).
Hohenlohe, P. A., Funk, W. C. & Rajora, O. P. Population genomics for wildlife conservation and management. Mol. Ecol. 30, 62–82, https://doi.org/10.1111/mec.15720 (2021).
Sambrook, J., Fritsch, E. R. & Maniatis, T. Molecular Cloning: A Laboratory Manual 2nd edn (Cold Spring Harbor Laboratory Press, 1989).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Laetsch, D. R. & Blaxter, M. L. BlobTools: Interrogation of genome. F1000Res. 6, 1287, https://doi.org/10.12688/f1000research.12232.1 (2017).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368, https://doi.org/10.1038/s41592-021-01101-x (2021).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075, https://doi.org/10.1093/bioinformatics/btt086 (2013).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single–copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Ou, S. & Jiang, N. LTR_retriever: A Highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422, https://doi.org/10.1104/pp.17.01310 (2017).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358, https://doi.org/10.1093/bioinformatics/bti1018 (2005).
Bao, W. et al. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 5, 4.10.11–14.10.14, https://doi.org/10.1002/0471250953.bi0410s05 (2004).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full–length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268, https://doi.org/10.1093/nar/gkm286 (2007).
Dobin, A. et al. STAR: ultrafast universal RNA–seq aligner. Bioinformatics 29, 15–21, https://doi.org/10.1093/bioinformatics/bts635 (2013).
Hoff, K. J., Lomsadze, A., Borodovsky, M. & Stanke, M. Whole–Genome Annotation with BRAKER. Methods Mol. Biol. 5, 65–95, https://doi.org/10.1007/978-1-4939-9173-0_5 (2019).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59, https://doi.org/10.1186/1471-2105-5-59 (2004).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript–level expression analysis of RNA–seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667, https://doi.org/10.1038/nprot.2016.095 (2016).
Altschul, S. F. et al. Gapped BLAST and PSI–BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402, https://doi.org/10.1093/nar/25.17.3389 (1997).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res. 14, 988–995, https://doi.org/10.1101/gr.1865504 (2004).
Grabherr, M. G. et al. Trinity: reconstructing a full–length transcriptome without a genome from RNA–Seq data. Nat. Biotechnol. 29, 644, https://doi.org/10.1038/nbt.1883 (2011).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA–seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512, https://doi.org/10.1038/nprot.2013.084 (2013).
Kent, W. J. BLAT–– The BLAST–like alignment tool. Genome Res. 12, 656–664, https://doi.org/10.1101/gr.229202 (2002).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–75, https://doi.org/10.1093/bioinformatics/bti310 (2005).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Lowe, T. M. & Chan, P. P. tRNAscan–SE On–line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57, https://doi.org/10.1093/nar/gkw413 (2016).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100–fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Marchler-Bauer, A. et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203, https://doi.org/10.1093/nar/gkw1129 (2017).
Thomas, P. D. et al. PANTHER: A Library of Protein Families and Subfamilies Indexed by Function. Genome Res. 13, 2129–2141, https://doi.org/10.1101/gr.772403 (2003).
Wilson, D. et al. SUPERFAMILY—Sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 37, D380–D386, https://doi.org/10.1093/nar/gkn762 (2009).
Lewis, T. E. et al. Gene3D: Extensive Prediction of Globular Domains in Proteins. Nucleic Acids Res. 46, D435–D439, https://doi.org/10.1093/nar/gkx1069 (2018).
Letunic, I. & Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496, https://doi.org/10.1093/nar/gkx922 (2018).
Hulo, N. et al. The 20 years of PROSITE. Nucleic Acids Res. 36, D245–D249, https://doi.org/10.1093/nar/gkm977 (2008).
Jones, P. et al. InterProScan 5: genome–scale protein function classification. Bioinformatics 30, 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 38, 5825–5829, https://doi.org/10.1093/molbev/msab293 (2021).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30, https://doi.org/10.1093/nar/28.1.27 (2000).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29, https://doi.org/10.1038/75556 (2000).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31645377 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBJUWB000000000 (2024).
Chen, X. & Han, W. Orange cup coral (Tubastraea coccinea) genome. Figshare https://doi.org/10.6084/m9.figshare.27987716 (2025).
Soares-Souza, G. B. et al. The genomes of invasive coral Tubastraea spp. (Dendrophylliidae) as tool for the development of biotechnological solutions. Preprint at https://doi.org/10.1101/2020.04.24.060574 (2020).
Acknowledgements
This research was funded by Innovational Fund for Scientific and Technological Personnel of Hainan Province (KJRC2023A02), Outstanding Talent Team Project of Hainan Province (HNYT20240001), Young Scientists Fund of the National Natural Science Foundation of China (42106117), and PI Project of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (GML20220018, YQ2024004). We also acknowledge the support of the High-Performance Biological Supercomputing Center at the Ocean University of China for this research.
Author information
Authors and Affiliations
Contributions
Z.B. and S.W. conceived and designed the study. Z.B. and S.W. coordinated and supervised the whole study. X.M.C. and W.H. conducted the genome assembly and analysis. X.Y.C. and Y.T. extracted DNA. K.C., L.B., L.Z. and J.H. participated in discussions and provided suggestions for manuscript improvement. Xiaomei Chen and W.H. did most of the writing with input from other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, X., Han, W., Chang, X. et al. High-quality genome assembly of the azooxanthellate coral Tubastraea coccinea (Lesson, 1829). Sci Data 12, 507 (2025). https://doi.org/10.1038/s41597-025-04839-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04839-7
