
Abstract
Hibiscus yunnanensis S.Y. Hu is an endangered species of the genus Hibiscus (Malvaceae), which has high potential economic value. However, the absence of a high-quality reference genome impedes the study of the ecology and molecular biology of H. yunnanensis. Here, we present a high-quality chromosome-level assembly of H. yunnanensis using BGI-DIPSEQ, Nanopore, and Hi-C sequencing. The assembled genome size is 2.2 Gb with a contig N50 of 12.1 Mb and a scaffold N50 of 137.1 Mb. Approximately 99.2% of the assembly is anchored into 17 pseudochromosomes, and a BUSCO analysis indicates a completeness score of 99.6%. Furthermore, we identify 42,085 protein-coding genes, of which 96.4% are functionally annotated. This genome resource provides a foundation for future studies on unique traits, including drought-tolerant, savanna-adapted, and long-flowering traits. Its ability to flower in winter, along with its automatic selfing and lack of delayed inbreeding depression, makes it an excellent model for studying style curvature mechanism and its adaptive significance in the Malvaceae.
Similar content being viewed by others

Background & Summary
Hibiscus is a genus of flowering plants belonging to the Malvaceae family, which encompasses over 300 species1. It is widely distributed in warm-temperate, tropical, and subtropical regions across the world and has relatively high economic value. These species are well-known for their large, colorful, and visually striking flowers, making them highly valued as ornamental plants. Certain species, such as H. syriacus (also known as Rose of Sharon) and H. rosa-sinensis (China rose), are extensively cultivated in gardens and landscapes for their aesthetic appeal2.
Besides its ornamental value, Hibiscus has long been utilized for diverse purposes. Studies have shown its significant medicinal properties, including antioxidant3,4, anti-inflammatory5,6, antitumor7, antihypertensive8, and antimicrobial activities9,10. The genus also has culinary applications, with flowers and other plant parts used in beverages such as hibiscus tea, which is globally popular for its refreshing flavor and health benefits, as well as in food products11. In addition, species such as H. tiliaceus provide fibers for textile production, including rope-making and construction materials12. Similarly, H. cannabinus is used in paper-making and supplies materials for crafting and construction13.
Hibiscus yunnanensis S.Y. Hu is a perennial shrub that primarily grows in subtropical regions on sunny, dry, and hot mountain slopes at elevations of 400–600 m. Since Hu (1955) established the species with Herry A. [13218] as the type specimen14, it has been found that H. yunnanensis is only distributed in Yuanjiang Hani, Yi and Dai Autonomous County, Yuxi City, Yunnan Province. It has been threatened and classified as endangered (EN)15 due to its restricted distribution, limited number of mature individuals, and declining habitat quality. It exhibits unique morphological characteristics, such as a yellow, bell-shaped corolla with a central purplish-red coloration (Fig. 1a), distinguishing it from the predominately white, pink, and red flowers of most other Hibiscus species. This distinct floral morphology imparts significant ornamental value, making H. yunnanensis a promising candidate for horticultural applications, including seed propagation and cultivation. Furthermore, it grows in dry and hot valleys, showing remarkable adaptation to high temperatures and limited water availability, with ecological characteristics resembling those of savannas, such as open landscapes, sparse vegetation, and seasonal climatic extremes16. Hibiscus yunnanensis has a long flowering period and can flower off-season in winter. Currently, no studies have specifically investigated the style curvature mechanism in H. yunnanensis. However, given its characteristic automatic selfing without inbreeding depression, we hypothesize that the species may utilize a unique style curvature mechanism to ensure reproductive success and prevent inbreeding depression. Therefore, a high-quality reference genome is important for promoting the comprehensive study of H. yunnanensis. Such genomic resources will facilitate the integration of genomic data with ecology, thereby enhancing our understanding and exploitation of this species.

Morphology and genomic features of H. yunnanensis. (a) Floral morphology from a frontal view, showing the characteristic yellow, bell-shaped corolla with a central purplish-red coloration. (b) Lateral view showing the calyx morphology along with trichomes, which can emit fetid smell to deter potential herbivores. (c) Circos plot of H. yunnanensis genome. The tracks from outside to inside display the chromosomes, gene number, GC content, Repeat density, LTR density, LTR/Copia density, LTR/Gypsy density, collinearity block of self-vs-self. (d) Hi-C interactive heat map.
Here, we present the genome of H. yunnanensis using ONT reads (263.7 Gb, 119.9×), NGS reads (315.4 Gb, 141.4×), Hi-C reads (210.8 Gb, 94.5×), and RNA-seq 134.3 Gb, 94.5×). The assembled contig size is close to the estimated genome size of 2.2 Gb based on k-mer estimates, with a scaffold N50 length of 137.1 Mb. A total of 99.2% of the assembled sequences are anchored to 17 pseudo-chromosomes. The genome contains 42,085 protein-coding genes, and 96.4% of them are annotated. The high-level genome assembly and annotation of H. yunnanensis will provide insights into the ecology within the genus Hibiscus, laying the foundation for ecological and molecular genetics studies.
Methods
Sampling
Hibiscus yunnanensis individuals were collected from Yuanjiang Hani, Yi, and Dai Autonomous County in Yuxi City, Yunnan Province. These plants were then self-pollinated to produce second-generation individuals in the greenhouse of Kunming Botanical Garden (Fig. 1a). Fresh young leaves from these second-generation plants were collected and stored for DNA extraction. Additionally, we collected young leaves, mature leaves, stems, fruits, budding flowers, full-blooming flowers, and roots from the same plant for transcriptome sequencing. Three biological replicates for each sample were immediately frozen in liquid nitrogen and subsequently stored at –80 °C.
Library construction and sequencing
We employed a modified CTAB (cetyltrimethylammonium bromide) method17 to extract high-quality genomic DNA from young H. yunnanensis leaves. The concentration of the extracted DNA was measured using both a Nanodrop (Nanodrop Technologies, Wilmington, DE, USA) and a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). To ensure the purity and integrity of the DNA, a 1% agarose gel electrophoresis was performed.
Genomic DNA fragments ranging from 200 to 400 base pairs (bp) were used for second-generation short-read library preparation. A total of 1 μg of genomic DNA was used following the manufacturer’s protocol provided by BGI. Short-read libraries were then subjected to paired-end (PE) sequencing using the BGI-DNBSEQ platform18 (BGI Inc., Shenzhen, China) with a PE 150 model, producing 315.4 Gb of raw data at approximately 141.4 × coverage (Table 1; Supplementary Table S2).
For ONT library preparation and sequencing19, the Nanopore DNA library was prepared using the SQK-LSK108 Kit from Oxford Nanopore Technologies (Oxford, UK). The sequencing of this library was performed on a Nanopore GridIONX5 sequencer using five flow cells. Base calling was carried out using Guppy v4.0.11 within the MinKNOW package, generating 151.2 Gb of data with roughly 67.8 × coverage for assembly (Table 1; Supplementary Table S3).
We used the TIANGEN kit with DNase I to extract total RNA, which was then prepared into a paired-end library with a 250 bp insert size using the NEBNextUltraTM RNA Library Prep Kit (Supplementary Table S5). These libraries were then sequenced on the BGI-DIPSEQ platform. Low-quality data was filtered out using Trimmomatic v0.3920 with the parameters ILLUMINACLIP:adapter.fa:2;30:10 LEADING:5TRAILING:5, generating 134.3 Gb of 100 bp paired-end data.
Hi-C library construction and sequencing
Hi-C library construction was carried out using the DpnII restriction enzyme and a method from the BGI QingDao Institute21. The chromatin was digested with DpnII and labeled at the ends with biotin-14-dATP (Thermo Fisher Scientific, Waltham, MA, USA). The DNA was then extracted, purified, and sheared using a Covaris S2 (Covaris, Woburn, MA, USA). Thus, prepared Hi-C libraries were sequenced on the BGI-DIPSEQ platform, producing approximately 210.8 Gb (94.5×) of data with 150 bp paired-end reads (Table 1; Supplementary Table S4).
Genome size estimation
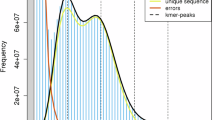
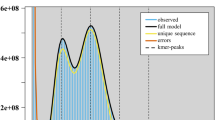
In order to estimate the genome size of the H. yunnanensis genome, k-mer spectral analysis of 60 × BGI-DIPSEQ short reads was utilized. The k-mer frequencies with a size of 17 were used to estimate the genome size from the short BGI-DIPSEQ reads. The 17-mer frequency distribution analysis, performed with GenomesScope222, estimated the H. yunnanensis genome to be 2.2 Gb. We applied strict quality control using SOAPfilter v2.223 to reduce sequencing errors. For assessing genome heterozygosity, variant calling on whole-genome short-read data was performed using the Genome Analysis Toolkit (GATK) v4.2.3.024, resulting in a heterozygosity value of 0.001%.
Genome assembly and optimization
For genome assembly, four different software and parameters were tested to optimize assembly quality. NextDenovo v2.225 was utilized with parameters read_cutoff = 1k, seed_cutoff = 32937, along with other default settings, processing both 100 × and 65 × nanopore data. Canu v2.026 was used with 65 × nanopore data, employing parameters-d./result merylThreads = 40 genomeSize = 2.32 g minReadLength = 1000 minOverlapLength = 500 corOutCoverage = 120 corMinCoverage = 2 and other defaults. Similarly, we used Wtdbg2 v2.527 to assemble the genome using parameters -x ont -g 2.4 g, utilizing 65 × nanopore data. Also, Flye v2.9.128 was used for assembly in three different configurations: ① using 65 × ONT data with a parameter of min-overlap 5000; ② using 65 × ONT data with a parameter of min-overlap 10,000; and ③ using 100 × ONT data with a parameter of min-overlap 10,000. By comparing the results of the different software and parameters, including coverage, BUSCO, and N50, the Flye ① assembly with a contig N50 value of 11.6 Mb was considered the primary contig genomes for subsequent analysis (Supplementary Table S6).
Subsequently, NextPolish v1.3.029 was utilized to refine the initial draft of assembled contigs (Flye ①) through six polishing rounds—two rounds with ONT long reads and four rounds with short reads. Following this, purge_dups v1.2.330 was employed to curate the contigs, generating scaffold N50 length of 12,097,940 bp (Table 1; Supplementary Table S7). This selection process considered the mapped read coverage obtained from short read data and alignments using Minimap231.
Hi-C paired-end reads were processed with Trimmomatic v0.3920 to remove low-quality bases and adapter sequences. To compute the contact frequency, all filtered reads were aligned to the contig assembly using Juicer v332. The 3D-DNA pipeline v18092233 was then executed with two iterative rounds to correct misjoining (-r2), while other with default parameters. Manual inspection and refinement of the draft assembly were performed using Juicebox v1.11.0834 (Fig. 1c).
Assessment of the assembled genome
We used BUSCO v3.0.235 to assess the quality and completeness of the assembled H. yunnanensis genome by comparing it against the embryophyta_odb10 dataset, capturing 99.6% of the 1,614 core eukaryote genes (Supplementary Table S8). Additionally, RNA reads were mapped to the draft assembly using Hisat2 v2.1.036, achieving a mapping rate of >95.1%. Whole genome sequence short reads were also mapped with BWA37, resulting in a 99.8% mapping rate and 99.1% coverage, which collectively indicates a high-quality genome assembly.
Repetitive elements identification
Both de novo and homology-based approaches were used to identify repeat sequences in the genome. The de novo approach involved constructing a novel repeat library using LTR_retriever v2.838, LTR_FINDER v1.0.739, and RepeatModeler240. Following this, we used RepeatMasker v4.0.841 to annotate the repeat elements. Tandem repeats were specifically identified using Tandem Repeats Finder v4.0742. For the homology-based approach, repeat elements were predicted through comparative analysis using RepeatMasker v4.0.8 and RepeatProteinMask v4-0-741. Repetitive elements in H. yunnanensis showed a moderate proportion of repetitive elements, comprising 73.2% of genome assembly, with LTR/Gypsy elements contributing 42.5% (Fig. 1b, Table 2).
Protein coding genes prediction
Protein-coding gene sets were predicted using de novo gene prediction, homology-based annotation, and transcriptome-based prediction methods. For the de novo approach, gene prediction was executed on a repeat-masked genome using Augustus v3.0.343, GlimmerHMM v3.0.244, and SNAP v11/29/201345. For homology-based gene prediction, amino acid sequences from Durio zibethinus, Gossypium raimondii, Theobroma cacao, and three related species (H. cannabinus, H. syriacus, and H. trionum) were compared using GeMoMa v1.3.146 and the UniProt database (release 2021_04). TBLASTN v2.2.18 (e-value cutoff: 1e-5)47 was employed to identify putative homologous genes by aligning protein sequences across the entire genome. Subsequently, GeneWise v2.2.048 was used to refine the alignment regions, providing accurate exon and intron information. In the RNA-seq-based gene prediction approach, clean RNA-seq reads were aligned to the assembled genomes using Hisat2 v2.0.436. Gene prediction was performed by identifying cDNAs through a genome-guided method with StringTie v1.2.249, followed by mapping these cDNAs back to the genome using PASA v2.3.350. The assembled cDNA sequences from Trinity v2.6.651 were then aligned to the H. yunnanensis genome sequences using BLAT v34 × 1252. Similarly, a non-redundant gene set was generated using maker v353 pipeline, resulting in the identification of 42,085 protein-coding genes. The distributions of mRNA length, CDS length, intron length, and exon number in H. yunnanensis align closely with those observed in other species genomes (Fig. 2), which supports the assembly of a high-quality genome for H. yunnanensis.

Comparison of the distribution of gene elements for each gene among seven representative species. (a) mRNA length. (b) CDS length. (c) Exon length. (d) Intron length. The x-axis represents the length (bp) and the y-axis represents the density of genes or exons or introns. The species compared are H. yunnanensis (HYUN), D. zibethinus (Dzib), G. raimondii (Grai), H. cannabinus (Hcan), H. syriacus (Hsyr), H. trionum (Htri), and T. cacao (Tcac).
Functional annotation
To functionally annotate the protein-coding genes, we performed sequence similarity and domain conservation analysis. BLASTP was used for the initial homolog search against public protein databases, with an e-value cutoff of 1e-5 and criteria including top hit 5, amino acid identity >0.3, and match length >0.5. The databases included SwissProt (release-2020_05)54, KEGG (59.3)55, TrEMBL (release-2020_05)54, and the NCBI non-redundant protein NR database (20201015). InterProScan v5.28-67.056 was then used to detect and classify domains and motifs, providing comprehensive functional annotations. The annotation rate for H. yunnanensis was found to be 96.4% (Table 3).
Data Records
The Nanopore, Hi-C, BGI-DIPSEQ, and RNA sequencing data used for genome assembly and annotation have been deposited in the Genome Sequence Archive (GSA) of the National Genomics Data Center (NGDC) under accession number CRA02220957. Additionally, all raw genomic sequencing data are available in the CNGB Nucleotide Sequence Archive (CNSA) under accession CNP000398558. The final contigs and chromosome assembly have been submitted to NCBI with the accession number of GCA_048544135.159. Annotation files, including predicted CDS and protein sequences as well as GFF files can be accessed on Figshare60. All other data produced or analyzed in this study are included within the article.
Technical Validation
We conducted an assessment of genome completeness using BUSCO v3.0.235, employing the embryophyta_odb10 database. Of the 1,614 core embryophyta genes, H. yunnanensis exhibited an identification rate of 99.6% (Table 1). To further validate the assembly’s completeness, we performed short-read mapping using clean raw data, where 99.8% of reads were properly paired with H. yunnanensis. We used Bridger tool61 to assemble the transcriptome sequences, followed by mapping to scaffold assemblies using BLAT52, yielding a pairing rate of 95.1%. Subsequently, BUSCO analysis was repeated after the Hi-C assembly, confirming similar results to those obtained from the ONT genome assemblies.
Code availability
All commands and pipelines used in data processing and analysis were performed according to the manual and protocols provided by the respective tools utilized in this study. The software and tools employed are openly accessible to the public, with the version and parameters mentioned in the Methods section (Supplementary Table S1). No specific or custom code was used in this study.
References
Hibiscus, L. https://powo.science.kew.org/taxon/urn:lsid:ipni.org:names:328182-2 (2024).
Brickell, C. The Royal Horticultural Society A-Z Encyclopedia of Garden Plants. (Dorling Kindersley, United Kingdom, 2008).
Da-Costa-Rocha, I., Bonnlaender, B., Sievers, H., Pischel, I. & Heinrich, M. Hibiscus sabdariffa L. – A phytochemical and pharmacological review. Food Chem. 165, 424–443 (2014).
Surana, A. R., Kumbhare, M. R., Gunjal, A. R., Goswami, S. S. & Ghuge, D. M. Chemical characterization, thrombolytic and antioxidant activity of Hibiscus tiliaceus L. leaves. Nat. Prod. Res. 36, 6106–6110 (2022).
Hamadjida, A. et al. Antioxidant and anti-inflammatory effects of Boswellia dalzielii and Hibiscus sabdariffa extracts in alloxan-induced diabetic rats. Metabol. Open 21, 100278 (2024).
Xu, X. Y., Tran, T. H. M., Perumalsamy, H., Sanjeevram, D. & Kim, Y.-J. Biosynthetic gold nanoparticles of Hibiscus syriacus L. callus potentiates anti-inflammation efficacy via an autophagy-dependent mechanism. Mater. Sci. Eng. C Mater. Biol. Appl. 124, 112035 (2021).
Nguyen, C. et al. Hibiscus flower extract selectively induces apoptosis in breast cancer cells and positively interacts with common chemotherapeutics. BMC Complement. Altern. Med. 19, 98 (2019).
Sanou, A. et al. In vivo diuretic activity and anti-hypertensive potential of Hibiscus sabdariffa extract by inhibition of angiotensin-converting enzyme and hypertension precursor enzymes. Foods 13, 534 (2024).
Baena-Santillán, E. S. et al. Comparison of the antimicrobial activity of Hibiscus sabdariffa calyx extracts, six commercial types of mouthwashes, and chlorhexidine on oral pathogenic bacteria, and the effect of Hibiscus sabdariffa extracts and chlorhexidine on permeability of the bacterial membrane. J. Med. Food 24, 67–76 (2021).
Portillo-Torres, L. A. et al. Antimicrobial effects of aqueous extract from calyces of Hibiscus sabdariffa in CD-1 mice infected with multidrug-resistant enterohemorrhagic Escherichia coli and Salmonella Typhimurium. J. Med. Food 25, 902–909 (2022).
Salem, M. A. et al. Hibiscus sabdariffa L.: phytoconstituents, nutritive, and pharmacological applications. Adv. Tradit. Med. 22, 497–507 (2022).
Surata, I. W., Nindhia, T. G. T. & Marsetio Widagdo, D. Promoting natural fiber from bark of Hibiscus tiliaceus as rope to reduce marine pollution from microplastic fiber yield from synthetic rope. E3S Web Conf. 158, 04007 (2020).
Jasmani, L. & Ainun, Z. M. A. Pulping and papermaking of kenaf. in Pulping and papermaking of nonwood plant fibers 143–155 (Elsevier, 2023).
Hu, S. Y. Flora of China Malvaceae [Fam. 153]. (The Arnold Arboretum of Harvard University, Cambridge, 1955).
Hibiscus yunnanensis. http://www.iplant.cn/rep/prot/Hibiscus%20yunnanensis.
Zhu, H., Tan, Y., Yan, L. & Liu, F. Flora of the savanna-like vegetation in hot dry valleys, Southwestern China with implications to their origin and evolution. Bot. Rev. 86, 281–297 (2020).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytochem. Bull. 19, 11–15 (1987).
Huang, J. et al. A reference human genome dataset of the BGISEQ-500 sequencer. Gigascience 6, 1–9 (2017).
Cherf, G. M. et al. Automated forward and reverse ratcheting of DNA in a nanopore at 5-Å precision. Nat. Biotechnol. 30, 344–348 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Hu, J. et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biol. 25, 107 (2024).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158 (2020).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Guiglielmoni, N., Houtain, A., Derzelle, A., Van Doninck, K. & Flot, J.-F. Overcoming uncollapsed haplotypes in long-read assemblies of non-model organisms. BMC Bioinformatics 22, 303 (2021).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. P. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinformatics 25, 4.10.1–4.10.14 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312 (2004).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89 (2016).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Kent, W. J. BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370 (2003).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–120 (2005).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA022209 (2025).
CNGB Nucleotide Sequence Archive https://db.cngb.org/search/project/CNP0003985/ (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_048544135.1 (2025).
Fang, D. Chromosome-level genome assembly of the threatened ornamental plant Hibiscus yunnanensis. Figshare https://doi.org/10.6084/m9.figshare.28114067.v1 (2025).
Chang, Z. et al. Bridger: a new framework for de novo transcriptome assembly using RNA-seq data. Genome Biol. 16, 30 (2015).
Acknowledgements
We are grateful to Fengmao Yang, Nuo Wu, and Zhi Chen for collecting samples. The work was supported by the Regional Innovative Development Joint Fund of NSFC to Hang Sun (U23A20149), the Key Project of Basic Research of Yunnan Province, China to Gao Chen (202301AS070001), and the National Key R&D Program of China to Tao Deng (2024YFF1306700).
Author information
Authors and Affiliations
Contributions
Bishal Gurung & Dongming Fang: writing original draft, visualization, methodology. Jiani Li, Dongming Fang and Qiongqiong Lin: investigation, resources, data curation, editing, formal analysis. Xing Guo & Gao Chen: conceptualization, review & editing, supervision, validation, funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gurung, B., Li, J., Fang, D. et al. Chromosome-level genome assembly of the threatened ornamental plant Hibiscus yunnanensis. Sci Data 12, 503 (2025). https://doi.org/10.1038/s41597-025-04842-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04842-y
This article is cited by
-
Advancing ornamental plant breeding through genomic technologies: opportunities, challenges, and future directions
Functional & Integrative Genomics (2025)
