
Abstract
The Gayal (Bos frontalis) is a rare semi-wild Bovine species that inhabits the harsh environments of Indo-China. Although the origins of the Gayal remain largely enigmatic, addressing the lack of comprehensive transcriptomic data is critical for understanding its genetic and molecular characteristics, which are essential for formulating effective conservation and management plans. In this study, an integrated PacBio Iso-seq and RNA-seq analysis was conducted on samples from 10 different organs and tissues of the Gayal, with each being sequenced in triplicate. The samples analyzed included the heart, liver, spleen, lung, kidney, rumen, abomasum, duodenum, ileum, and rectum. This comprehensive analysis resulted in the identification of 30,760 full-length transcripts ranging from 363 bp to 7,157 bp, with transcript information matched to seven commonly used databases. Gene family clustering and phylogenetic analyses encompassed a comprehensive dataset of 9 Bovine species, including the Gayal. Additionally, long non-coding RNAs (lncRNAs) were identified across all sampled tissues, and comprehensive gene expression profiles and differential expression gene analyses were performed. These findings provide a rich repository of genetic information, laying the foundation for comprehensive functional genomics studies and paving the way for deeper insights into the molecular mechanisms of the Gayal, thereby advancing our understanding of its transcriptome architecture and offering crucial data for conservation efforts and practical applications.
Similar content being viewed by others

Background & Summary
The Gayal, also known as Mithun (Bos frontalis), is a rare semi-wild Bovine species found in the harsh environments of Indo-China1,2. In China, the Gayal is predominantly located in the narrow valleys of the Dulong and Nujiang Rivers, as well as the adjacent mountainous areas of Yunnan Province, where it is referred to as ‘Dulong cattle’3. The Gayal has a chromosome complement of 2n = 58, which is exactly intermediate between that of domesticated cattle (Bos taurus, 2n = 60) and wild gaur (Bos gaurus, 2n = 56)4,5,6. This unique animal is larger in body size than local counterparts and is characterized by distinctive ‘white stockings’ on its stout legs7. Additionally, the Gayal exhibits prominent beef-type characteristics and has high-quality carcass attributes, particularly with a Longissimus dorsi muscle fiber diameter that is nearly half the size of that found in most other Bos species8,9,10. Because of the isolation of its habitats and a range of ecological and sociopolitical challenges, the Gayal is one of the least studied ungulates11. It is classified as a threatened species by the International Union for Conservation of Nature (IUCN). There is still significant uncertainty about its origins, and information on the transcriptome architecture of the Gayal is notably sparse. Addressing this knowledge gap is crucial for gaining a comprehensive understanding of its genetic and molecular characteristics, which is essential for devising effective conservation and management strategies.
Transcriptomic analysis serves as a powerful tool, playing a pivotal role in uncovering gene expression, function, and regulatory mechanisms12,13,14. Recent transcriptomic studies of Bos species among the Gayal, yak (Bos grunniens), and cattle, have provided profound insights into their gene expression profiles, metabolic pathways, and adaptive characteristics15. Such information not only enhances our understanding of how these species survive in harsh environments but also reveals their unique adaptations to specific ecological niches. Moreover, comparative analysis of the Gayal transcriptome with that of other related species can elucidate its evolutionary history and genetic relationships, thereby providing a scientific basis for conservation and restoration efforts.
In this study, samples were extracted from 10 organs and tissues of the Gayal, including the heart, liver, spleen, lung, kidney, rumen, abomasum, duodenum, ileum, and rectum, followed by an integrated PacBio Iso-seq and RNA-seq analysis. This resulted in the identification of 30,760 full-length transcripts, with lengths ranging from 363 bp to 7,157 bp. Through meticulous bioinformatics analysis, it was found that 15,783 and 14,064 of these transcript sequences matched entries in the NCBI non-redundant protein (NR) and Swiss-Prot databases, respectively. Further annotation indicated that 14,690, 13,609, and 11,406 transcripts had corresponding entries in the Kyoto Encyclopedia of Genes and Genomes (KEGG), euKaryotic Ortholog Groups (KOG), and Gene Ontology (GO) databases, respectively. Additionally, long non-coding RNAs (lncRNAs) were identified across all sampled tissues of the Gayal. These discovered full-length transcripts and gene expression profiles constitute a rich repository of genetic information, providing a foundation for comprehensive functional genomics studies and paving the way for deeper insights into the molecular mechanisms of the Gayal.
Methods
Ethics statement
All Gayal handling and experimental procedures in this study were approved by the Animal Care and Use Committee of Yunnan Agricultural University (Approval Code: 202307018, Approval Data: 3 July 2023).
Collection of samples and preparation of RNA
A 3-year-old male Gayal was sacrificed and samples were collected from the Yunnan Provincial Dulong cattle conservation and breeding base of Fenghuang Mountain, Lushui City, China. The tissue samples, including heart, liver, spleen, lung, kidney, rumen, abomasum, duodenum, ileum, and rectum, were promptly preserved in liquid nitrogen. The extracted RNA samples were first added to a DNase I buffer, followed by the addition of an appropriate amount of DNase I enzyme. The RNA samples underwent DNase I digestion at 37 °C for 30 minutes. DNase I stop solution was then added to ensure complete removal of the DNase I enzyme. Finally, the RNA samples were purified and assessed to ensure the absence of residual DNA. RNA was extracted from each tissue using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. Initial evaluation of RNA concentration and purity was performed using a Nanodrop 2000, while agarose gel electrophoresis was employed to check for genomic contamination, purity, and RNA integrity. Subsequently, the RNA integrity number was precisely determined using the Agilent 2100 platform.
PacBio library construction and sequencing
For PacBio library construction, RNA samples from 10 different organs and tissues were mixed in equal proportions. The library preparation and sequencing were performed at Wuhan Frasergen Gene Biotechnology Co., Ltd (Wuhan, China) using the PacBio Sequel II platform with a total of 6 SMRT cells. The 3′ ends of eukaryotic mRNAs feature a poly-A tail, and primers with Oligo dT were paired with the poly-A tail to synthesize cDNA via reverse transcription. These primers were added to the ends of the full-length cDNA synthesized through reverse transcription. The full-length cDNA was then amplified by PCR, and the products were purified using PB magnetic beads to remove cDNA fragments smaller than 1 kb. The ends were repaired and SMRTbell adapters were ligated. Unligated fragments were digested with exonucleases, and the sequencing library was purified using PB magnetic beads. After library construction, accurate quantification was performed using Qubit 3.0, and the library size was assessed using an Agilent 2100. Sequencing was only conducted if the library size met the expected criteria. The raw data obtained from the sequencer were filtered and processed using SMRTlink 10.0 software, with the parameter–minLength = 50 to exclude sequences shorter than 50 bp. After this filtering process, the resulting data were considered valid.
Illuminal mRNA library construction and sequencing
mRNA was isolated from 10 different organs and tissues of the Gayal using Oligo(dT) magnetic beads. Total RNA was extracted using TRIzol reagent (Invitrogen, CA, USA), and the purity and integrity of the RNA were monitored using a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). A 1.5% agarose gel was used to assess RNA contamination. mRNA purification was conducted according to the manufacturer’s instructions, followed by library construction using the V AHTS Universal V6 RNA-seq Library Kit for MGI (Vazyme, Nanjing, China). The quality and size of the libraries were evaluated using a Qubit 2.0 fluorometer (Life Technologies, Carlsbad, CA, USA) and the Agilent 2100 system (Agilent Technologies, CA, USA). Subsequently, sequencing was performed on a MGI-SEQ 2000 platform by the Wuhan Frasergen Bioinformatics Co., Ltd. (Wuhan, China). The RNA-seq experiments were performed with three biological replicates.
PacBio ISO-seq data processing
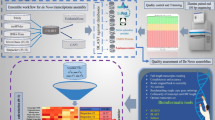
As shown in Table 1, a total of 32,505,744 subreads were generated from PacBio ISO-seq. The pbccs tool (https://github.com/nlhepler/pbccs) was employed to generate Circular Consensus Sequences (CCS) from subreads. To retain low-frequency transcripts and maximize data recovery, the “--all” parameter was used, resulting in 629,246 CCS reads, which were converted to FASTQ format for quality assessment using FastQC. Subsequent processing with the lima tool (https://lima.how) removed adapter sequences, primers, and barcode tags, yielding 366,942 full-length non-chimeric (FLNC) reads with an average length of 1,899 bp. The FLNC reads were further refined using the IsoSeq3 refine module to eliminate poly-A tails and chimeric sequences, producing a high-confidence dataset suitable for clustering. To generate consistent transcript clusters, the refined FLNC reads were processed using the IsoSeq3 cluster module, which removed redundancies and aggregated similar sequences. The resulting clustered transcripts underwent error correction with LoRDEC16 (https://gitlab.inria.fr/serge.lordec/lordec), a tool that utilizes high-accuracy short-read sequencing data for base-level corrections. After correction, the dataset was reduced to 30,760 high-quality isoforms with an average length of 1,904 bp and an accuracy exceeding 99% (Fig. 1A). This non-redundant full-length transcript dataset, which was generated through deduplication or clustering methods, ensures that each sequence or cluster represents a unique gene, meaning that the sequences or clusters in the dataset are non-redundant, with each corresponding to a distinct gene. This approach is consistent with the general definition of Unigene, where the non-redundant sequences or clusters provide a solid foundation for downstream analyses.

Isoform analysis and annotation of the Gayal’s ISO-seq data. (A) Matrix layout for all intersections of seven databases sorted by size. Dark circles in the matrix indicate sets that are part of the intersection. (B) Venn diagram analysis of ISO-seq isoforms with NR, KOG, COG, Swiss-Prot, EggNOG, GO, and KEGG databases. (C) KOG functional analysis of ISO-seq isoforms.
Function annotation of unigenes
Functional annotation of the Gayal Unigenes, a non-redundant full-length transcript dataset obtained through deduplication or clustering, was conducted through detailed comparisons with seven databases (NR, NT, Swiss-Prot, GO, KEGG, KOG, and COG). For the NT database analysis, BLAST software was used with an E-value cutoff of <1e-10. Annotation for NR, KOG, COG, and Swiss-Prot was performed using the Diamond BLASTX method17, with the same E-value threshold of <1e-10. GO annotations for the Gayal transcriptome were derived from NR database alignments using the Diamond BLASTX tool. The BLAST output was processed to extract UniProt IDs, which were subsequently mapped to GO terms using the UniProt idmapping.tb.gz file. A custom Python script, Uniprot2GO_annotated.py, was utilized to match UniProt IDs with GO terms based on the mapping file. The script outputs a comprehensive association of query sequences with GO terms, providing functional insights into the transcriptome. KEGG functional classification was comprehensively analyzed using the KofamKOALA method (https://www.genome.jp/tools/kofamkoala/).
Quality control of annotation
To ensure the quality of annotations, the full-length transcripts were comprehensively checked against multiple reference databases. Matches were found in the following databases: 26,758 (86.99%) in Nr database, 23,629 (76.82%) in KOG, 24,064 (78.23%) in Swiss-Prot, 25,161 (81.80%) in EggNOG, and 6,118 (19.89%) in COG. Additionally, 17,256 (56.10%) and 18,825 (61.20%) transcripts were annotated in the KEGG and GO databases, respectively (Fig. 1B). Further functional classification of the isoforms was performed using the KOG database (Fig. 1C). Analysis of GO ontology and KEGG pathways was conducted using TBtools-II18 to identify annotated sequences and explore active biological pathways in the Gayal (Fig. 2). For example, KEGG analysis revealed that Unigenes are primarily involved in six pathways: metabolism, genetic information processing, environmental information processing, cellular processes, organismal systems, and human diseases. Additionally, the coding sequences of the Gayal were predicted and analyzed using TransDecoder software.

GO ontology and KEGG pathways analysis for the annotated sequences of the Gayal.
In the sequence similarity-based clustering analysis (Fig. 3A), we utilized protein sequences from 9 Bovine species, including the Gayal. All alignments were performed with an E-value threshold of <1e−5. A total of 22,963 genes were identified in the Gayal, revealing multiple homologous gene clusters shared with the other 8 Bovine species. Subsequently, a phylogenetic tree was constructed using maximum likelihood methods with OrthoFinder19 (Fig. 3B). The analysis demonstrated that the Gayal exhibits a close phylogenetic relationship with the Bos javanicus species. These phylogenetic relationships align with the known taxonomy and evolutionary positions of these species.

Cluster analysis of gene families and phylogenetic analysis. Cluster analysis of gene families of 9 Bovine species and phylogenetic analysis of 9 Bovine species though a rooted STAG species phylogenetic tree using the maximum likelihood method.
Identification of open reading frames (ORFs) and long non-coding RNAs (lncRNAs)
ORFs were predicted in the Gayal transcripts using the TransDecoder v2.0.1 software package (https://transdecoder.github.io/). Transcripts with complete ORFs, along with 5′ and 3′ untranslated regions, were identified as full-length transcripts. lncRNAs play crucial roles in the regulation of growth and development in many Bovine species. Potential lncRNAs were predicted using CNCI (Coding-Non-Coding Index)20, CPC2 (Coding Potential Calculator 2)21, CPAT (Coding Potential Assessment Tool)22, and PLEK (Prediction of Long Non-coding RNAs)23 software tools.
For CNCI, the CNCI.py script was run with the “ve” model for vertebrate species using the following command: python /path/to/CNCI.py -f /path/to/transcripts.fa -o CNCI_output -p 8 -m ve.
For CPC2, the command was: python /path/to/CPC2.py -i /path/to/transcripts.fa -o CPC2_output.
For CPAT, the following command was used: python /path/to/cpat.py -g /path/to/transcripts.fa -d /path/to/ Mouse_logitModel.RData -x /path/to/ Mouse_Hexamer.tab -o CPAT_output.
For PLEK, the command was: python /path/to/PLEK.py -fasta /path/to/transcripts.fa -out PLEK_output -thread 20. Figure 4 presents a Venn diagram analysis of the lncRNA predictions from these four software tools, revealing a total of 17,565 identified lncRNAs.

(A) Venn diagram analysis of predicted lncRNAs based on ISO-seq transcripts. (B) predicted lncRNAs number of four tools.
Identification of differentially expressed genes across tissues
Gene expression levels across the 10 organs and tissues of the Gayal were first determined through a correlation analysis and by calculating the RPKM (Reads Per Kilobase of transcript per Million mapped reads) values. To identify Differentially Expressed Genes (DEGs), we applied a log-fold change (log FC) threshold of >2 or <-2, with a false discovery rate (FDR) <0.001 and a p-value < 0.005 to ensure statistical significance.
First, we performed quality control on the raw RNA-seq data using tools: FastQC, MultiQC, Trimmomatic. We used FastQC for initial quality assessment and Trimmomatic for trimming low-quality reads and adapters from the raw data. For paired-end sequencing data, Trimmomatic was used with parameters to perform adapter clipping, quality trimming, and filtering short reads. The cleaned reads from the 10 tissue samples were aligned to the reference isoforms generated from PacBio ISO-seq data using Bowtie2. After alignment, we performed gene expression quantification using featureCounts, followed by DEG analysis using DESeq 2 (or edgeR), which is widely used for RNA-seq data analysis. The resulting DEG list was filtered based on the aforementioned thresholds for log-fold change, FDR, and p-value to identify genes with significant differential expression across the tissues. Subsequent correlation analysis (Fig. 5A) and gene expression profile examination in 10 organs and tissues (Fig. 5B) provided in-depth insights into the dynamic mRNA expression of the Gayal.

(A) Gene expression profile in 10 organs and tissues (heart, liver, spleen, lung, kidney, rumen, abomasum, duodenum, ileum, and rectum) in the Gayal. (B) Heatmap analysis of the differentially expressed mRNAs in 10 organs and tissues.
Cluster analysis of gene families and phylogenetic analysis
In the gene family clustering analysis, we utilized protein sequences from 9 Bovine species (Bos taurus, Bos indicus, Bos indicus × Bos taurus, Bos grunniens, Bos javanicus, Bubalus bubalis, Ovis aries, Capra hircus). Redundant and alternatively spliced sequences were removed, retaining only the longest transcripts. Multiple sequence alignments were performed using the “-S diamond” option, and a phylogenetic tree was constructed based on maximum likelihood methods using the “-M msa -T iqtree” parameters, resulting in a STAG (Species Tree Inference for All Genes) phylogenetic tree.
Data Records
In this study, all raw RNA-seq data for the Gayal have been deposited in the Sequence Read Archive (SRA) at the National Center for Biotechnology Information, under the accession number SRR30184652, SRR30184649, SRR30184653, SRR30184654, SRR30184655, SRR30184656, SRR30184657, SRR30184658, SRR30184659, SRR30184651, SRR30184632, SRR30184631, SRR30184648, SRR30184629, SRR30184647, SRR30184646, SRR30184645, SRR30184644, SRR30184643, SRR30184642, SRR30184641, SRR30184650, SRR30184640, SRR30184639, SRR30184638, SRR30184637, SRR30184636, SRR30184635, SRR30184630, SRR30184634, SRR3018463324,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53. The all assemblies (assembled transcriptome data), transcriptome annotation, and read counts files have been uploaded to Zenodo database and can be accessed at https://doi.org/10.5281/zenodo.14859252. The data of all the analyses described in the methods have been deposited in the GEO repository, and the records have been assigned GSM accession numbers as GSE289623, GSM8795261, GSM8795262, GSM8795263, GSM8795264, GSM8795265, GSM8795266, GSM8795267, GSM8795268, GSM8795269, GSM8795270, GSM8795271, GSM8795272, GSM8795273, GSM8795274, GSM8795275, GSM8795276, GSM8795277, GSM8795278, GSM8795279, GSM8795280, GSM8795281, GSM8795282, GSM8795283, GSM8795284, GSM8795285, GSM8795286, GSM8795287, GSM8795288, GSM8795289, GSM879529054,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84.
Technical Validation
To reveal the global mRNA expression profile of the Gayal, sequencing was performed on samples from 10 organs and tissues (heart, liver, spleen, lung, kidney, rumen, abomasum, duodenum, ileum, rectum). The quality of the RNA-seq cleaned reads was assessed using FastQC, showing the average Q20 and Q30 values were 99.04% and 97.06%, respectively. The GC content distribution of Gayal tissue samples exhibited a normal pattern, indicating no sequencing data contamination (Fig. 6).

The sequence quality of the Gayal RNA-seq cleaned reads from 10 organs and tissues with three biological replicates.
Code availability
No custom script was used in this work. All software used in this study is publicly available, with parameters specified in the Methods section. Default settings recommended by the developers were used where specific parameters were not detailed.
References
Rajkhowa, S., Sarma, D. K. & Rajkhowa, C. Seroprevalence of Toxoplasma gondii antibodies in captive mithuns (Bos frontalis) from India. Veterinary Parasitology 135, 369–374 (2006).
Deng, W. et al. Phylogenetic Analysis of 16S rDNA Sequences Manifest Rumen Bacterial Diversity in Gayals (Bos frontalis) Fed Fresh Bamboo Leaves and Twigs (Sinarumdinaria). Asian-Australas. J. Anim. Sci. 20, 1057–1066 (2007).
Huaming, M. The biology characteristics of Gayal (Bos frontalis) and potential exploitation and utilization. Journal of Yunnan Agricultural University 20, 258–261 (2005).
Bhambhani, R. & Kuspira, J. The somatic karyotypes of American bison and domestic cattle. Can. J. Genet. Cytol. 11, 243–249 (1969).
Gallagher, D. S. Jr. & Womack, J. E. Chromosome conservation in the Bovidae. Journal of Heredity 83, 287–298 (1992).
Chi, J. et al. New insights into the karyotypic relationships of Chinese muntjac (Muntiacus reevesi), forest musk deer (Moschus berezovskii) and gayal (Bos frontalis). Cytogene. Genome Res. 108, 310–316 (2005).
Mukherjee, S. et al. Whole genome sequence and de novo assembly revealed genomic architecture of Indian Mithun (Bos frontalis). BMC Genomics 20, 617 (2019).
Xi, D. et al. Polymorphisms of the insulin-like growth factor-binding protein 3 gene (IGFBP3) in gayal (Bos frontalis). Gene 497, 98–102 (2012).
Li, Y. et al. Large-Scale Chromosomal Changes Lead to Genome-Level Expression Alterations, Environmental Adaptation, and Speciation in the Gayal (Bos frontalis). Mol. Biol. Evol. 40 (2023).
Ge, C., Tian, Y., Chen, T. & Wu, Y. Studies on the meat feature of gayal (Bos frontalis). Scientia Agricultura Sinica 29, 75–78 (1996).
Xi, D. et al. Genetic variability of the coding region for the prion protein gene (PRNP) in gayal (Bos frontalis). Mol. Biol. Rep. 39, 2011–2020 (2012).
Glinos, D. A. et al. Transcriptome variation in human tissues revealed by long-read sequencing. Nature 608, 353–359 (2022).
Drown, M. K., Crawford, D. L. & Oleksiak, M. F. Transcriptomic analysis provides insights into molecular mechanisms of thermal physiology. BMC Genomics 23, 421 (2022).
Chen, J. et al. Comparative transcriptomic analysis provides insights into the genetic networks regulating oil differential production in oil crops. BMC Biol. 22, 110 (2024).
Ma, J. et al. Comparative transcriptome analyses of gayal (Bos frontalis), yak (Bos grunniens), and cattle (Bos taurus) reveal the high-altitude adaptation. Front. Genet. 12, 778788 (2022).
Salmela, L. & Rivals, E. LoRDEC: accurate and efficient long read error correction. Bioinformatics 30, 3506–3514 (2014).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Chen, C. et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 16, 1733–1742 (2023).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Sun, L. et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41, e166–e166 (2013).
Kong, L. et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349 (2007).
Wang, L. et al. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 41, e74–e74 (2013).
Li, A., Zhang, J. & Zhou, Z. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinf. 15, 1–10 (2014).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184652 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184649 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184653 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184654 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184655 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184656 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184657 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184658 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184651 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184632 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184631 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184648 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184629 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184647 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184646 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184645 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184644 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184643 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184642 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184641 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184650 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184640 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184639 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184638 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184637 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184636 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184635 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184630 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184634 (2024).
Sequence Read Archive (SRA) of the National Center for Biotechnology Information https://identifiers.org/ncbi/insdc.sra:SRR30184633 (2024).
NCBI GEO https://identifiers.org/geo/GSE289623 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795261 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795262 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795263 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795264 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795265 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795266 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795267 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795268 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795269 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795270 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795271 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795272 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795273 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795274 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795275 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795276 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795277 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795278 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795279 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795280 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795281 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795282 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795283 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795284 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795285 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795286 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795287 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795288 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795289 (2025).
NCBI GEO https://identifiers.org/geo/GSM8795290 (2025).
Acknowledgements
This work was supported by grants from the Major Science and Technology Projects in Yunnan Province: 202202AE090005, Yunnan Provincial Agricultural Union Foundation: 202401BD070001-008, and the “Xingdian Talent” Industry Innovation Talent Program in Yunnan Province (XDYC-CYCX-2022-0029).
Author information
Authors and Affiliations
Contributions
The conception and design of the study were led by W.D. and D.X.; Y.C., Y.L., J.H., J.W., M.L. were responsible for sample collection and total RNA isolation; Z.G. and B.W. conducted the bioinformatics analysis; Z.G. and B.W. drafted the manuscript, which was subsequently revised by W.D.; The final manuscript was read, reviewed, and approved by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gao, Z., Wang, B., Lu, Y. et al. De novo transcriptome assembly and annotation of the semi-wild Gayal (Bos frontalis). Sci Data 12, 589 (2025). https://doi.org/10.1038/s41597-025-04927-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04927-8
