
Abstract
Microbial communities can vary as a function of seasonal precipitation and the phenotypic characteristics of the prevailing plant species in an ecosystem. The Brazilian campos rupestres (CRs) host a unique flora adapted to harsh conditions, including severe droughts and nutrient-poor soils. Velloziaceae, a dominant angiosperm family in CRs, exhibit contrasting drought adaptive strategies, prominently desiccation tolerance and dehydration avoidance. Here, we created a comprehensive dataset of microbial composition and dynamics of bulk soil and distinct plant compartments (leaf blade, dry sheath, aerial root, and underground root) from two desiccation-tolerant and two dehydration-avoiding, non-desiccation-tolerant Vellozia species, across four seasons (beginning and end of rainy and dry seasons) through 16S rRNA gene sequencing of 374 samples. This dataset also includes 38 soil metagenomes encompassing dry and rainy seasons from both drought adaptive strategies. Exploring an overlooked aspect of CRs biology offers significant potential for understanding plant-microbial associations and adaptations to water availability in tropical regions. The genetic data and metadata support further research for hypothesis testing and cross-study comparisons.
Similar content being viewed by others

Background & Summary
The response of microbial communities to seasonal precipitation is not the same across different ecosystems. While changes are reported for environments such as temperate forests, and agricultural and aquatic ecosystems1,2,3,4, in semiarid and desert grasslands, seasonal precipitation does not always modify richness and evenness of soil microbial communities5,6 or soil microbial attributes such as biomass and respiration7. The phenotypic characteristics of the plant species that prevail in each environment also contribute to shape the composition of bacterial communities8,9.
The campos rupestres (CRs; rupestrian grasslands) are a disjunct montane ecoregion in Eastern Brazil featuring rocky outcrops, sandy and stony soils, and diverse flora including grasses, sedges, shrubs and small trees10,11. The flora of CRs has evolved to withstand harsh conditions characterised by severe seasonal droughts and soil nutritional deficiencies, particularly phosphorus, in addition to other stresses10,11. Within the CRs, the Velloziaceae stands out as an iconic, dominant, and species-rich angiosperm family11,12,13,14,15,16, with species of its major genera Vellozia and Barbacenia showcasing contrasting adaptive strategies to drought that range from dehydration avoidance to desiccation tolerance17,18. While dehydration-avoiding, non-desiccation-tolerant (NDT) species can sustain high relative water content (RWC) during drought stress, desiccation-tolerant (DT) species can tolerate RWC as low as 5% and, upon rehydration, quickly resume the same metabolism and photosynthetic rates as before desiccation17,18,19.
Although the CRs cover less than 1% of Brazil’s continental area, this megadiverse habitat hosts around 15% of the country’s flora, with approximately 40% of these species being endemic, resulting in the highest endemism-to-area ratio in Brazil11,12,13,14,15,20. The CRs share physical and functional characteristics with the fynbos of the Cape Floristic Region and the kwongan of the Southwestern Australia Floristic Region21. Despite the high plant species richness and endemism in these regions, their microbial communities are poorly characterised. Few and recent studies have explored the structure of fynbos and kwongan bacterial communities22,23,24,25, and, in the CRs, only the microbiomes associated with the Velloziaceae species B. macrantha and V. epidendroides, that differ in phosphorus acquisition strategies and substrates they live on (rocks and sandy soils, respectively), have been characterised26,27. However, to our knowledge, no studies have shown whether microbial community attributes in those megadiverse regions respond to seasonal variations in rainfall or different drought adaptive strategies of host plants, with a particular lack of data for the CRs.
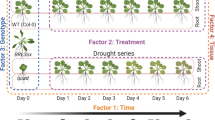
In this study, to address whether these factors help shape microbial communities, we collected multiple samples, during dry and rainy seasons, from individuals of four Vellozia species with contrasting drought adaptive strategies: two DT species, V. nivea L. B. Sm. & Ayensu and V. tubiflora (A. Rich.) Kunth, and two NDT species, V. intermedia Seub. and V. peripherica Mello-Silva17,18,28 (Fig. 1), as well as associated soil samples. We used 16S rRNA gene sequencing to characterise microbiomes in host plant compartments (i.e., leaf blade, dry sheath, aerial root, and underground root) and bulk soil. The study experimental design is shown in Fig. 2. This dataset (Dataset 129) can provide valuable insights into microbiome composition and dynamics throughout dry and rainy seasons and host compartments of Vellozia species with contrasting drought adaptive strategies.

Physiognomy of the Vellozia species investigated in this study. (a,b), V. nivea; (c,d), V. tubiflora; (e,f), V. intermedia; (g,h), V. peripherica. (a,c,e,g), individuals during rainy seasons; (b,d,f,h), individuals during dry seasons.

Summary of the experimental design. (a) Schematic depiction of the seasons, plant drought adaptations, and the sampled compartments used in the study. (b) Data analysis process workflow.
Sampling of soil and plant tissue was performed from September 2018 through September 2019 during four distinct seasons (Fig. 3): early rainy (ER), late rainy (LR), early dry (ED), and late dry (LD) seasons. We measured the moisture of leaves of DT and NDT individuals and their associated soils with the aim of correlating water status imposed by precipitation variation with microbial composition. Soil moisture content varied significantly (Fig. 4a), being ∼ 80 times lower in the LD season compared to the LR season. Soils associated with DT species displayed 3–7 times lower moisture content during the ED and LD seasons than those with NDT species (Fig. 4a). NDT species sustained high leaf moisture content across all seasons (97–100% in ER and LR seasons and 94–97% in ED and LD seasons), while DT species exhibited a significant decrease in leaf moisture during the LD season (80–100% in the ER and LR seasons, 80–90% in ED season, and ∼15% in the LD season) (Fig. 4b).

Meteorological data, and soil and plant sampling in the CRs study sites. Daily maximum, mean and minimum temperatures and daily precipitation were obtained from the Global Precipitation Measurement (GPM) mission satellite data for the nearest coordinates to the study sites.

Moisture content in different seasons. (a) Soil moisture content. (b) Leaf moisture content. Letters indicate significance (p < 0.05) on ANOVA followed by Tukey HSD.
A total of 374 samples were used to produce high-throughput sequencing data of DNA fragments amplified from the 16S V3-V4 rRNA gene regions. Processing these data retrieved an average of 503,325 reads per sample and identified 257,790 unique amplicon sequence variants (ASVs). Additionally, we detected 47 identified prokaryotic phyla. The common phyla across all surveyed groups, with a relative abundance exceeding 3%, included Proteobacteria, Acidobacteriota, Planctomycetota, Actinobacteriota, and Chloroflexi (Fig. 5).

Vellozia microbial community composition at the phylum level. The horizontal bars at the top of the graph categorize samples by compartment (Leaf blade, Dry sheath, Aerial root, Underground root, and Bulk soil). Directly below, color-coded bars categorize samples according to drought adaptation strategies: the desiccation-tolerant (DT) species V. nivea and V. tubiflora are represented in orange, and the non-desiccation-tolerant (NDT) species V. intermedia and V. peripherica in green. At the bottom, rectangles denote seasons: Early rainy (light blue), Late rainy (dark blue), Early dry (orange), and Late dry (red). The ‘Others’ category includes phyla with mean relative abundances below 3%.
The diversity among sample groups was evident, with soil displaying the highest diversity (p < 0.01), followed by underground roots. No significant differences were observed for dry sheath and aerial root; however, the leaf blade stood out as the least diverse sample (p < 0.01) (Fig. 6).

Comparisons of species richness and abundance distribution. (a) Across plant compartments and bulk soil samples in five distinct sample types: Leaf blade (Lb), Dry sheath (Dsh), Aerial root (Ar), Underground root (Ur), and Bulk soil (S); (b) between desiccation-tolerant (DT) and non-desiccation-tolerant (NDT) drought-adaptive strategies; and (c) across Early rainy (ER), Late rainy (LR), Early dry (ED), and Late dry (LD) seasons. Comparisons were calculated using the Shannon index. Differences between samples were assessed using the Wilcoxon test, with a significance threshold of p < 0.05. The resulting p-values were adjusted using the Benjamini-Hochberg correction method.
The reported data also comprise shotgun metagenomic data from soil associated with all DT and NDT species during both dry and rainy seasons for functional assessment of microbial communities. Total DNA extracted from the Vellozia-associated soil underwent shotgun sequencing, resulting in a total of 1.5 TB and 687.7 GB for rainy and dry season samples, respectively. Samples from rainy seasons were assembled individually, resulting in 19 metagenomes with an average assembly length of 122,471,070 bp, an average contig number of 82,179, and a mean N50 of 1,367 bp. Similarly, samples from dry seasons were also assembled individually, producing 19 metagenomes with an average assembly length of 30,074,623 bp, an average contig number of 20,997, and a mean N50 of 1,347 bp (Dataset 229).
Annotation of these metagenomes retrieved a median number of 510 noncoding genes and 74,965 protein-coding genes. In total, we identified 478 pathways, 5,158 KOs, and 2,190 enzyme categories across 38 soil metagenome samples associated with Vellozia species. Our comparison revealed 82 MetaCyc pathways that were differentially abundant (FDR < 0.05) between the dry and rainy seasons (Dataset 329). Additionally, we found 63 pathways that were differentially abundant between communities associated with DT and NDT survival strategies (FDR < 0.05).
This study represents an initial effort to (i) explore the associated microbiome dynamics across different seasons in the megadiverse Brazilian CRs and (ii) identify microorganisms associated with the contrasting desiccation tolerance and dehydration avoidance drought adaptive strategies exhibited by members of the outstanding Velloziaceae family. This study explores an overlooked aspect of CRs biology and offers significant potential for understanding plant-microbial associations and adaptations to conditions of water seasonal availability in tropical regions. This first-time published dataset enables reuse and supports further hypothesis testing and cross-study comparisons.
Methods
Sample collection for DNA extraction
Specimens of two DT species (V. nivea and V. tubiflora) and two NDT species (V. intermedia and V. peripherica) belonged to large populations located within the Serra da Canastra National Park (V. peripherica and V. tubiflora) and a private property adjacent to the Park (V. intermedia and V. nivea), respectively in the municipalities of São Roque de Minas and Delfinópolis, Minas Gerais state, Brazil. Coordinates and elevation (in m.a.s.l.) of the respective CRs sites are: V. nivea, S 20° 29′15.8″ W 46° 32′05.3″, 950; V. tubiflora, S 20° 13′50.8″ W 46° 37′31.2″, 1,380; V. intermedia, S 20° 29′07.2″ W 46° 30′49.8″ 1,002; V. peripherica, S 20° 15′36.9″ W 46° 25′22.0″, 1,355. These sites exhibit typical CRs features (Fig. S1), with quartzite outcrops and shallow, sandy gravel soil.
The Serra da Canastra region in southern Minas Gerais state experiences a humid subtropical climate (Koppen’s Cwb)30 characterised by a rainy period from October to March (spring and summer) and a dry period from April to September (fall and winter). Field samplings were performed from September 2018 through September 2019 during four distinct seasons: early rainy (ER) season in September 2018, late rainy (LR) season in February and March 2019, early dry (ED) season in June 2019, and late dry (LD) season in August and September 2019 (Fig. 3). Global Precipitation Measurement (GPM) mission satellite data for the virtual station TRMM.1570, which has the nearest coordinates (20°30′00.0“S, 46°30′00.0“W) to all study sites, were retrieved from the Agritempo agrometeorological monitoring system (http://www.agritempo.gov.br/).
To obtain representative samples of microbial communities associated with Vellozia plants, in each season five similarly sized individuals of each species were selected from approximately 400-m² plots. Consequently, 20 individuals were sampled for each species across four seasons. Individuals were pried out from soil, preserving a large portion of their fasciculate root system as a single clod, and organs were dissected from individuals. Plant samples encompassed several organs: both young and mature leaf blades, dry sheaths enveloping the apical, median, and basal segments of the “pseudostem” as well as aerial and underground roots of varying diameters. Attached soil was gently shaken from underground roots. Soil samples for DNA extraction were obtained by pooling material excavated at a 15 cm depth from two diametrically opposite areas located at 20 cm from the central stem of each individual. All samples were stored in conical tubes and immediately placed in dry ice without prior washing. Each sampling event yielded approximately 50 ml of soil or fragments of plant materials for DNA extraction. Subsequently, the collected samples were stored at −80 °C in the laboratory until further processing.
Leaf and soil sampling and moisture analyses
For soil moisture analysis, soil was sampled at a 15-cm depth from two diametrically opposite areas located at 20 cm from the central stem of each plant. The two samples from each individual were pooled, and soil moisture content was determined using 50 g through convective oven-drying thermogravimetry31 at 105 °C until constant weight was achieved.
Leaf relative moisture content was determined through standing rehydration, as described by Arndt et al.32, on medium-sized leaves from two branches of each individual (totalling 64 samples). In short, leaf blades stored in tightly sealed plastic vials on ice were weighted within 24 hours after sampling for estimating fresh mass. Leaves were placed into 50-ml tubes with the cut end submerged in distilled water. Water-saturated mass and dry mass were measured by sequentially weighting leaves after imbibition for 24 h at 4 °C and drying at 75 °C for approximately 48 h, respectively.
Statistical analysis of soil and leaf moisture contents was performed using ANOVA followed by Tukey HSD (p < 0.05) in R33. All moisture analyses were performed at the Instituto Agronômico de Campinas (Campinas, Brazil).
DNA extraction and 16S ribosomal RNA (rRNA) gene sequencing
Microbial diversity was assessed through the sequencing of 16S rRNA gene amplicons, molecular markers for bacterial taxonomy34,35,36. This analysis utilised microbial DNA derived from both soil and Vellozia plant organs collected in the four seasons (ER, LR, ED, and LD). Plant samples underwent cryogenic pulverisation under liquid nitrogen without any pre-treatment using the cryogenic mills models 6970EFM and 6870 (SPEX SamplePrep, Metuchen, NJ, USA) at the Centro Nacional de Pesquisa em Energia e Materiais (Campinas, Brazil) and the Analytical Chemistry Laboratory at Centro de Energia Nuclear na Agricultura (Piracicaba, Brazil), respectively. Pulverised samples were subsequently stored at −80 °C until further processing.
DNA extractions were performed with 250 mg of soil and 150 mg of pulverised plant samples using the DNeasy PowerSoil kit (Qiagen, Valencia, CA, USA), following the manufacturer’s protocol with the inclusion of a heating step at 65 °C for 10 minutes after the addition of solution C1. The extracted DNA was quantified using fluorimetry with the Qubit dsDNA BR Assay kit (Thermo Fisher Scientific Inc., MA, USA), and a NanoDrop spectrophotometer (Thermo Fisher Scientific). Extracted samples ranged in DNA concentrations from 5 ng/µl to 150 ng/µl. Quality was assessed by checking the 260/280 and 260/230 nm ratios and verifying the absence of non-specific bands through agarose gel electrophoresis.
Amplicon library targeted the V3-V4 regions of the 16S rRNA gene specific to prokaryotes, using the V3-338f 5′-ACWYCTRCGGGRGGCWGCA-3′ and V4-806r 5′-GGACTACHVGGGTWTCTAAT-3′ primers37, with chloroplast and mitochondrial Peptide Nucleic Acid (PNA) clamp oligos38. The samples were sequenced on the Novaseq 6000 SP500 v1.5 platform (Illumina, San Diego, CA, USA), generating paired-end reads of 2 × 250 bp, at the Donnelly Sequencing Centre (University of Toronto, Toronto, Canada).
Shotgun metagenomic sequencing and assembly
DNA extracted from soil samples collected during the LR and LD seasons for the four species was employed for shotgun metagenomic sequencing on the Novaseq 6000 SP500 v1.5 platform (Illumina) at the Donnelly Sequencing Centre, resulting in paired-end reads of 2 × 250 bp. The raw sequencing data were quality filtered using the JGI RQCFilter pipeline v39.06 (https://sourceforge.net/projects/bbmap/). This pipeline removes adapters and filters out reads shorter than 51 bp or less than 33% of their original length. It also trims bases with quality scores below 10 from both sequence ends. Additionally, the pipeline performs cross-contamination filtering, eliminating short contaminants, pJET1 sequences, and other unwanted sequences, including common microbial and human DNA contamination. Processed sequences were assembled using MEGAHIT v1.2.9 with parameters --k-list 23,43,63,83,103,123 and --min-contig-len 100039. Assembly quality statistics were subsequently generated using QUAST v5.2.0 --mgm --min-identity 90.040.
The structural and functional annotation, including the prediction of CRISPR elements, non-coding genes, protein-coding genes, and the assignment of gene functions, was conducted using the IMG Annotation Pipeline v.5.2.141. Quantitative profiles of microbial metabolic pathways and other molecular functions from metagenomic samples were generated using HuMAnN 342 (http://huttenhower.sph.harvard.edu/humann). This was followed by an analysis of pathway distinctions between the DT and NDT strategies, as well as between rainy and dry seasons, using the MaAsLin2 R package43 with default settings.
Microbial community profiling
We used fastp to remove adapters, trim low-quality bases with a quality score lower than 20, and discard reads shorter than 50 bp44. Filtered FASTQ sequences were imported into Qiime2 version 2023.945 for primer removal using the cutadapt plugin and subsequent processing through the DADA2 pipeline46. This pipeline included filtering, dereplicating, denoising, merging, removing chimeras, and grouping sequences into amplicon sequence variants (ASVs). The resulting DADA2-processed data is available as Dataset 429. Taxonomic assignment of ASVs was conducted using the Qiime2-feature-classifier plugin and the classify-sklearn47 function, utilising the Silva 138 99% OTUs database48. Sequences assigned as mitochondria, chloroplast, Eukaryote, and Unassigned were subsequently removed. Additionally, singletons—ASVs detected only once—were excluded. Samples with sequence numbers lower than 1000 were filtered out for the Vellozia microbiome composition analysis.
The data were exported to generate a phyloseq object49, and subsequent analysis was conducted using the microbiome package50 with R33. In the diversity analysis, we rarefied the sequences to a depth of 5,000 without replacement. The alpha diversity was represented by the Shannon index on microbiome package. We used the paired Wilcoxon test as the statistical method, with a p-value threshold set at 0.05. The resulting p-values were then adjusted using the Benjamini-Hochberg correction method51.
Data Records
Raw data of both the 16S rRNA gene amplicon sequencing and the shotgun sequencing were deposited in the NCBI Sequence Read Archive SRP51276052. BioProject, sample description, SRA Study, and SRA Run accessions of each of the sequencing libraries generated in this study are available in Dataset 529. Shotgun sequencing assembly files are available through the Joint Genome Institute Genome OnLine Database53 GOLD ID Gs0166663 and European Nucleotide Archive PRJEB8808154.
To support reanalysis by other researchers, we have deposited Dataset 629 (Processed Amplicon Data) that contains ASV identifiers, taxonomic classification, taxonomic classification confidence, ASV DNA sequences, and ASV abundance for each sample.
Datasets 1–6 can be accessed on Figshare29 (https://doi.org/10.6084/m9.figshare.28389302). Details on the data contained in each file are as follows:
Dataset 1. Sample information for this study
This dataset contains information of Library type, 16S rRNA gene amplicon or Shotgun metagenome; Drought adaptative strategy, Desiccation-tolerant (DT) or Non-desiccation-tolerant (NDT); Species, Vellozia nivea (Vn), Vellozia tubiflora (Vt), Vellozia intermedia (Vi), and Vellozia peripherica (Vp); Sample material, Leaf blade (Lb), Dry sheath (Dsh), Aerial root (Ar), Underground root (Ur), and Bulk soil (S)); Sampling month (September, February, June, and August), Season (Early rainy, Late rainy, Early dry, and Late dry); sample #, number of samples; Sequencing, sequencing direction and sequence length; and NCBI sample name.
Dataset 2. Metagenome assembly and annotation statistics
This dataset contains IDs for ENA analysis, NCBI library names and Gold Biosample; Assembly metrics such as Assembly length (bp); Number of contigs; N50; L50; Max scaffold length (bp); Metagenome Annotation counts for RNA genes (rRNA genes, 5S rRNA, 16S rRNA, 18S rRNA, 23S rRNA, 28S rRNA, tRNA genes); Protein coding genes; Noncoding genes (with Product Name, with COG, with Pfam, with KO, with Enzyme, with MetaCyc, with KEGG, COG clusters, Pfam clusters); and CRISPR count.
Dataset 3. Comparison of MetaCyc pathways between rainy and dry seasons, as well as NDT and DT drought adaptive strategies
This dataset contains Differentially abundant metabolic pathways; metadata, compared variable (season or drought adaptative strategy); value, the reference feature; coef, model coefficient value for the reference feature; stderr, the standard error from the model; N, number of samples in the statistical comparison; N.not.0, total number of samples in which the feature is non-zero; pval, nominal significance of this association; and qval, significance corrected with Benjamini-Hochberg method51.
Dataset 4. Summary of sequence progression through DADA2 steps
This dataset contains NCBI library ID; sample-id, NCBI sample ID; input, number of input sequences; number and percentage of input passed filter sequences; number of denoised sequences, number and percentage of input merged sequences; number and percentage of input non-chimeric sequences.
Dataset 5. NCBI accessions of all shotgun and amplicon sequencing samples generated in the study
This dataset contains NCBI SRA Run; Download path, Experiment ID; Library Name; Library Strategy, Metagenome shotgun sequencing or amplicon sequencing; Model, sequencing platform; SRA Study ID; BioProject ID; Sample ID, BioSample ID, Submission ID; sample Description; Gold Biosample ID; and ENA analysis ID.
Dataset 6. Processed amplicon dataset
This dataset contains Feature ID, ASV identifiers; Taxon, taxonomic classification; Confidence, taxonomic classification confidence; Sequence, ASV DNA sequences; and ASV abundance for each sample.
Technical Validation
The quality and purity of the extracted DNA was assessed using fluorimetry with the Qubit dsDNA BR Assay kit (Thermo Fisher Scientific) and spectrophotometry with the NanoDrop spectrophotometer (Thermo Fisher Scientific) by checking the 260/280 and 260/230 nm ratios. DNA concentration in the extracted samples ranged from 5 ng/µl to 150 ng/µl. PCR of the 16S rRNA gene was verified for the absence of non-specific bands through agarose gel electrophoresis. The prepared libraries were quantified using the NEBNext Library Quant pPCR kit (New England Biolabs, Ipswich, Massachusetts, USA).
Read quality assessment revealed that most sequences scored Q25 or higher, indicating high base call accuracy. DADA2 quality control and denoising of 16S rRNA genes ensure that each DNA sequence variation arises from biological mutations rather than sequencing errors. The ASVs have an average length of 407.84 bp, with a maximum length of 438 bp and a standard deviation of 38.71 bp, indicating complete length of the V3-V4 16S rRNA gene region, enhancing taxonomic classification resolution.
The variation of microbial composition across different organs and soil revealed the expected higher sample relatedness with closer spatial proximity. While bulk soil samples exhibited greater similarity to underground root samples, aerial root samples were more similar to dry sheath samples, and leaf blade samples were the most distinct (Fig. 5). These observations evidence no technical issues with sample preparation and analysis.
Code availability
The codes used to process and analyse the 16S rRNA gene amplicon and shotgun metagenome data from the Vellozia microbiome are available on GitHub (https://github.com/GCCRC-dev/VelloziaMicrobiome).
References
Liu, Y. et al. Rainfall-induced changes in aquatic microbial communities and stability of dissolved organic matter: Insight from a Fen river analysis. Environ. Res. J. 246, 118107, https://doi.org/10.1016/j.envres.2024.118107 (2024).
Raczka, N. C., Carrara, J. E. & Brzostek, E. R. Plant–microbial responses to reduced precipitation depend on tree species in a temperate forest. Glob. Change Biol. 28, 5820–5830, https://doi.org/10.1111/gcb.16340 (2022).
Yan, G. et al. Variations of the effects of reduced precipitation and N addition on microbial diversity among different seasons in a temperate forest. Appl. Soil Ecol. 166, 103995, https://doi.org/10.1016/j.apsoil.2021.103995 (2021).
Zhang, G. et al. Seasonality and assembly of soil microbial communities in coastal salt marshes invaded by a perennial grass. J. Environ. Manage. 331, 117247, https://doi.org/10.1016/j.jenvman.2023.117247 (2023).
Bell, C. W. et al. Soil microbial and nutrient responses to 7 years of seasonally altered precipitation in a Chihuahuan Desert grassland. Glob. Change Biol. 20, 1657–1673, https://doi.org/10.1111/gcb.12418 (2014).
Li, X., Yan, Y., Lu, X., Fu, L. & Liu, Y. Responses of soil bacterial communities to precipitation change in the semi-arid alpine grassland of Northern Tibet. Front. Plant Sci. 13, 1036369, https://doi.org/10.3389/fpls.2022.1036369 (2022).
Toledo, S., Gargaglione, V., Yahdjian, L. & Peri, P. L. Differential responses of soil microorganisms to precipitation changes in austral semiarid grasslands. Pedobiologia 97-98, 150873, https://doi.org/10.1016/j.pedobi.2023.150873 (2023).
Oyserman, B. O. et al. Disentangling the genetic basis of rhizosphere microbiome assembly in tomato. Nat. Commun. 13, 3228, https://doi.org/10.1038/s41467-022-30849-9 (2022).
Pérez-Jaramillo, J. E. et al. Linking rhizosphere microbiome composition of wild and domesticated Phaseolus vulgaris to genotypic and root phenotypic traits. ISME J. 11, 2244–2257, https://doi.org/10.1038/ismej.2017.85 (2017).
Oliveira, R. S. et al. Mineral nutrition of campos rupestres plant species on contrasting nutrient‐impoverished soil types. New Phytol. 205, 1183–1194, https://doi.org/10.1111/nph.13175 (2015).
Silveira, F. A. O. et al. Ecology and evolution of plant diversity in the endangered campo rupestre: a neglected conservation priority. Plant Soil 403, 129–152, https://doi.org/10.1007/s11104-015-2637-8 (2016).
Brazil Flora Group. Brazilian Flora 2020: Leveraging the power of a collaborative scientific network. Taxon 71, 178–198, https://doi.org/10.1002/tax.12640 (2022).
Colli‐Silva, M., Vasconcelos, T. N. C. & Pirani, J. R. Outstanding plant endemism levels strongly support the recognition of campo rupestre provinces in mountaintops of eastern South America. J. Biogeogr. 46, 1723–1733, https://doi.org/10.1111/jbi.13585 (2019).
Conceição, A. A., Pirani, J. R. & Meirelles, S. T. Floristics, structure and soil of insular vegetation in four quartzite-sandstone outcrops of “Chapada Diamantina”, Northeast Brazil. Braz. J. Bot. 30, 641–656, https://doi.org/10.1590/S0100-84042007000400009 (2007).
Conceição, A. A. et al. in Ecology and Conservation of Mountaintop grasslands in Brazil (eds Fernandes, G. W.) Ch. 6, 105–127 (Springer International Publishing, 2016).
Le Stradic, S., Buisson, E. & Fernandes, G. W. Vegetation composition and structure of some Neotropical mountain grasslands in Brazil. J. Mt. Sci. 12, 864–877, https://doi.org/10.1007/s11629-013-2866-3 (2015).
Alcântara, S. et al. Carbon assimilation and habitat segregation in resurrection plants: a comparison between desiccation‐ and non‐desiccation‐tolerant species of Neotropical Velloziaceae (Pandanales). Funct. Ecol. 29, 1499–1512, https://doi.org/10.1111/1365-2435.12462 (2015).
Teodoro, G. S. et al. Desiccation tolerance implies costs to productivity but allows survival under extreme drought conditions in Velloziaceae species in campos rupestres. Environ. Exp. Bot. 189, 104556, https://doi.org/10.1016/j.envexpbot.2021.104556 (2021).
Oliver, M. J. et al. Desiccation Tolerance: Avoiding Cellular Damage During Drying and Rehydration. Annu. Rev. Plant Biol. 71, 435–460, https://doi.org/10.1146/annurev-arplant-071219-105542 (2020).
Brazil Flora Group. Growing knowledge: an overview of Seed Plant diversity in Brazil. Rodriguésia 66, 1085–1113, https://doi.org/10.1590/2175-7860201566411 (2015).
Mucina, L. Vegetation of Brazilian campos rupestres on siliceous substrates and their global analogues. Plant life on campo rupestre, a megadiverse Neotropical old-growth grassland 238, 11–23, https://doi.org/10.1016/j.flora.2017.06.007 (2018).
Keet, J.-H., Ellis, A. G., Hui, C. & Le Roux, J. J. Strong spatial and temporal turnover of soil bacterial communities in South Africa’s hyperdiverse fynbos biome. Soil Biol. Biochem. 136, 107541, https://doi.org/10.1016/j.soilbio.2019.107541 (2019).
Teste, F. P. et al. Plant-soil feedback and the maintenance of diversity in Mediterranean-climate shrublands. Science 355, 173–176, https://doi.org/10.1126/science.aai8291 (2017).
Turner, B. L. et al. Contrasting patterns of plant and microbial diversity during long‐term ecosystem development. J. Ecol. 107, 606–621, https://doi.org/10.1111/1365-2745.13127 (2019).
Mpai, T., Jaiswal, S. K., Cupido, C. N. & Dakora, F. D. Seasonal Effect on Bacterial Communities Associated with the Rhizospheres of Polhillia, Wiborgia and Wiborgiella Species in the Cape Fynbos, South Africa. Microorganisms 10, 1992, https://doi.org/10.3390/microorganisms10101992 (2022).
Camargo, A. P. et al. Microbiomes of Velloziaceae from phosphorus-impoverished soils of the campos rupestres, a biodiversity hotspot. Sci. Data 6, 140, https://doi.org/10.1038/s41597-019-0141-3 (2019).
Camargo, A. P. et al. Plant microbiomes harbor potential to promote nutrient turnover in impoverished substrates of a Brazilian biodiversity hotspot. ISME J. 17, 354–370, https://doi.org/10.1038/s41396-022-01345-1 (2023).
De Freitas Larocca, P., Saldanha Mancio, J., Padilha, P., Mello-Silva, R. & Alcantara, S. Recent divergence in functional traits affects rates of speciation in the Neotropical Velloziaceae (Pandanales). Bot. J. Linn. 199, 144–172, https://doi.org/10.1093/botlinnean/boab102 (2022).
Pinto, O. H. B. et al. Seasonal bacterial profiles of Vellozia with distinct drought adaptations in the megadiverse campos rupestres. Figshare https://doi.org/10.6084/m9.figshare.28389302 (2025).
Alvares, C. A., Stape, J. L., Sentelhas, P. C., Gonçalves, J. L. d. M. & Sparovek, G. Köppen’s climate classification map for Brazil. Meteorol. Z. 22, 711–728, https://doi.org/10.1127/0941-2948/2013/0507 (2013).
Clarke Topp, G. & Ferré, P. A. in Methods of Soil Analysis Part 4 Physical Methods (eds Dane, J. H. & Topp, G. C.) Ch. 3, 417–545 (Soil Science Society of America, Inc., 2002).
Arndt, S. K., Irawan, A. & Sanders, G. J. Apoplastic water fraction and rehydration techniques introduce significant errors in measurements of relative water content and osmotic potential in plant leaves. Physiol. Plant. 155, 355–368, https://doi.org/10.1111/ppl.12380 (2015).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2021).
Mullis, K. B. & Faloona, F. A. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 155, 335–350, https://doi.org/10.1016/0076-6879(87)55023-6 (1987).
Woese, C. R. et al. Conservation of primary structure in 16S ribosomal RNA. Nature 254, 83–86, https://doi.org/10.1038/254083a0 (1975).
Lundberg, D. S., Yourstone, S., Mieczkowski, P., Jones, C. D. & Dangl, J. L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 10, 999–1002, https://doi.org/10.1038/nmeth.2634 (2013).
Liu, J. H., Zhang, M. L., Zhang, R. Y., Zhu, W. Y. & Mao, S. Y. Comparative studies of the composition of bacterial microbiota associated with the ruminal content, ruminal epithelium and in the faeces of lactating dairy cows. Microb. Biotechnol. 9, 257–268, https://doi.org/10.1111/1751-7915.12345 (2016).
Nielsen, P. E. & Egholm, M. An Introduction to Peptide Nucleic Acid. Curr. Issues Mol. Biol. 1, 89–104, https://doi.org/10.21775/cimb.001.089 (1999).
Li, D., Liu, C.-M., Luo, R., Sadakane, K. & Lam, T.-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676, https://doi.org/10.1093/bioinformatics/btv033 (2015).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075, https://doi.org/10.1093/bioinformatics/btt086 (2013).
Huntemann, M. et al. The standard operating procedure of the DOE-JGI Metagenome Annotation Pipeline (MAP v.4). Stand. Genomic. Sci. 11, 17, https://doi.org/10.1186/s40793-016-0138-x (2016).
Beghini, F. et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 10 https://doi.org/10.7554/eLife.65088 (2021).
Mallick, H. et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 17, e1009442, https://doi.org/10.1371/journal.pcbi.1009442 (2021).
Chen, S. Ultrafast one‐pass FASTQ data preprocessing, quality control, and deduplication using fastp. Imeta 2, e107, https://doi.org/10.1002/imt2.107 (2023).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857, https://doi.org/10.1038/s41587-019-0209-9 (2019).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583, https://doi.org/10.1038/nmeth.3869 (2016).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90, https://doi.org/10.1186/s40168-018-0470-z (2018).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596, https://doi.org/10.1093/nar/gks1219 (2012).
McMurdie, P. J. & Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 8, e61217, https://doi.org/10.1371/journal.pone.0061217 (2013).
Lahti, L. & Shetty, S. microbiome R package. Bioconductor https://doi.org/10.18129/B9.bioc.microbiome (2017).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300, https://doi.org/10.1111/j.2517-6161.1995.tb02031.x (1995).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP512760 (2024).
Mukherjee, S. et al. Twenty-five years of Genomes OnLine Database (GOLD): data updates and new features in v.9. Nucleic Acids Res. 51, D957–D963, https://doi.org/10.1093/nar/gkac974 (2022).
European Nucleotide Archive. https://identifiers.org/ena.embl:PRJEB88081 (2025).
Acknowledgements
The authors thank Recanto Ecológico Vale do Céu for granting access to CRs sites. This work was supported by grant 2016/23218-0 “Genomics for Climate Change Research Center” from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP). O.H.B.P. received a technical training fellowship (2022/08797-4) from FAPESP. B.B.B. received a graduate scholarship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). R.S.C.d.S. received a postdoctoral fellowship (2018/19100-9) from FAPESP.
Author information
Authors and Affiliations
Contributions
R.S.C.d.S. and I.R.G. conceived the project. J.E.C.T.Y., P.A., R.A.D., and I.R.G. acquired funding for the project. R.S.C.d.S. and B.B.B. performed soil and plant sampling. B.B.B. performed DNA extraction, PCR and sample preparation for sequencing and physicochemical analyses. O.H.B.P. performed bioinformatics analyses and data presentation. O.H.B.P., B.B.B., R.A.D. and I.R.G. wrote the manuscript with input from R.S.C.d.S., J.E.C.T.Y, and P.A. All authors read and approved the final manuscript. O.H.B.P. and B.B.B. contributed equally to this study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pinto, O.H.B., Biazotti, B.B., de Souza, R.S.C. et al. Seasonal bacterial profiles of Vellozia with distinct drought adaptations in the megadiverse campos rupestres. Sci Data 12, 636 (2025). https://doi.org/10.1038/s41597-025-04984-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04984-z
