
Abstract
The interdigital webs of aquatic tetrapods are a key structure evolved for adaptation, which are formed by inhibiting interdigital cell death (ICD). Diverse interdigital morphologies have independently evolved among species, and the regulatory mechanisms responsible for their development are still not fully understood. The Chinese soft-shell turtle (Pelodiscus sinensis) serves as a good research model that exhibits transitional traits from webless to fully webbed. In this study, we collected eight samples of interdigital webs from the fore- and hindlimbs of turtles at embryonic stage 19 (TK19) for RNA sequencing (RNA-seq) analysis. We identified 608 differentially expressed genes (DEGs). Whole-mount in situ hybridization (WISH) and real-time quantitative PCR (RT-qPCR) of representative genes confirmed the accuracy of the transcriptomic results. These findings not only provide new perspectives and data to support comparative studies of adaptive convergent evolution in aquatic animals but also enhance our understanding of the mechanisms underlying tetrapod limb morphogenesis. Furthermore, these results provide potential molecular targets for research on the plasticity of programmed cell death or senescence.
Similar content being viewed by others


Background & Summary
Webbed feet are a functional structure for the adaptation of aquatic tetrapods to aquatic life and show parallels to the evolution of forelimb wing membranes for flight in bats. Compared to mice, chickens and lizards, mammalian bats and dolphins, aquatic birds and the reptilian Chinese soft-shell turtle display weaker to no levels of cell death in their interdigital regions during embryonic development1,2,3,4,5, with the lack of cell death considered to be the basis for the formation of the interdigit webbing. The molecular mechanisms for interdigital cell death (ICD) have been extensively studied, with the retinoic acid (RA)6 and BMP7 signaling pathways, loss of cell and/or cell matrix adhesion8,9, and cellular aging pathways9 being shown to be involved. In contrast, much less information is known about the pathways antagonizing ICD, which would allow interdigit webs to form, and whether the same or different mechanisms are used among the different species. For example, in bats and dolphins, Fgfs and Gremlin work together to inhibit the BMP-mediated apoptosis in the interdigital region, while in ducks and Chinese soft-shell turtles, Fgfs are not involved2,5. Moreover, no obvious asymmetry has been observed in the expression patterns for gremlin among the webbed fore- and hindlimbs of Chinese soft-shelled turtles during embryonic development5, thus, it is difficult to determine whether the same anti-apoptotic mechanism is used by the Chinese soft-shell turtle.
In the absence of high-throughput technologies, previous studies mostly used the “candidate-gene” approach to identify genes involved in cell fate and interdigit webbing in waterfowl. However, the vast phylogenetic diversity of birds increases the difficulty of studying the development of interdigit webbing. Phylogenetic studies have shown that the Chinese soft-shell turtle diverged prior to the radiation of birds10, and its early evolution of webbed structures could make it a better suited model for Evo-Devo adaptation studies. As an important reptile in both aquatic and terrestrial ecosystems, its limb morphology shows a transitional pattern between terrestrial and aquatic life. During development from birth to adult, the interdigital webbing between the first and second phalanges (W1) largely regresses, the interdigital webbing between the second and third phalanges (W2) regresses approximately 50%. However, the weak distal ICD of the interdigital webbing between the third and fourth phalanges (W3) and between the fourth and fifth phalanges (W4) does not cause obvious regression and are thus nearly fully retained (Fig. 1). Similar patterns are observed for both the fore- and hindlimbs. Significant differences in webbing morphology and ICD can be observed during stage 19 (TK19) of embryonic development11. Given these differences, we therefore chose this developmental stage to identify differences in gene expression, which might identify molecular characteristics that regulate the differential fates of interdigital cells and explain the differences in webbing between the different digits of Chinese soft-shell turtles.

Morphology of the Chinese soft-shell turtle and the workflow for RNA-seq. (a) Morphology of a Chinese soft-shell turtle one month after hatching. The position of digit I is marked by an asterisk, bar = 1 cm. (b) Schematic diagram of the sampling for transcriptome sequencing (left) and the complete analytical workflow (right). The red lines in the schematics of the limbs on the left indicate the boundaries of the sampling range, with the green areas representing the regions where ICD occurs at TK195.
In this study, we dissected and separated the four interdigital membranes between the five digits of the forelimbs (FW1-4) and the hindlimbs (HW1-4) under a microscope. Total RNA was extracted from these samples for pool-sequencing. Each pool contained the paired limbs from 20 independent individuals (40 limbs). Sample collection areas and the RNA-seq workflow are shown in Fig. 1b. Given that the levels of cell death in the corresponding regions of the fore- and hindlimbs are very similar5, they could be considered to be biological replicates for studying web formation in the screening process. A total of 199.10 M reads were obtained by RNA-seq and the proportion of Q30 (phred quality score ≥ 30) reads in the clean data was over 93% in all samples. Hierarchical clustering analysis (HCA), based on the Spearman correlation coefficient, and three-dimensional principal component analysis (PCA), based on TPM (transcripts per million) data, both separated the samples into independent clusters according to whether they underwent apoptosis. The data distribution was consistent with the morphological characteristics, suggesting that this data can be effectively used for the screening of differential molecular features. Subsequently, we identified 608 differentially expressed genes (DEGs) with significant differences in expression. The accuracy and reliability of these results were verified by WISH and RT-qPCR of representative DEGs. In summary, our data reveal distinct molecular expression profiles of apoptotic and retained interdigital cells that not only provide a high-throughput data foundation for interspecies comparative studies on the development and evolution of webbed limbs, but also potentially identify molecular targets for cellular anti-apoptosis and anti-aging.
Methods
Sample collection
All animal procedures were approved by the Institutional Animal Care and Use Committee of Shenyang Agricultural University (approval ID: 202106038). Fertilized Chinese soft-shell turtle eggs (Pelodiscus sinensis) used in this study were obtained from Huayu aquaculture farm in Jizhou (Tianjin, China) and incubated in wet sand at 30–32 °C. Embryonic development was staged using the Tokita-Kuratani (TK) staging method to standardize the developmental process11. At developmental stage TK19, 20 morphologically consistent individuals were selected, and the four interdigital regions of the paired fore- and hindlimbs were dissected under a microscope (Leica, M165FC). Since the regression patterns of the interdigital webbing are conserved within the species and do not exhibit sexual dimorphism, we randomly selected the 20 embryonic individuals and did not record the sex of the samples. A total of eight sample groups were collected for pool-sequencing, with each group consisting of 40 tissues from left and right sides of the 20 individuals. The RNeasy Plus Mini Kit (Qiagen) was employed to extract total RNA. Subsequently, the purity and concentration of the total RNA was examined using Qubit (Life Technologies, Carlsbad, USA) and Nanodrop (Thermo, Waltham, USA). Moreover, the Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA) was adopted to assess the integrity of the RNA. The 260/280 nm ratio of all samples was between 2.06 and 2.11, and the RNA integrity number (RIN) values were all higher than 8.6, with the 28S/18S ribosomal RNA ratio ranging from 2.13 to 2.39 (Table 1).
RNA sequencing and quality control
A total of 3 μg of RNA per sample was used as the input material for library construction. Libraries were commercially sequenced (Biomarker Technologies Co., Beijing, China) using the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA). Each library yielded ~ 25 million 150 bp paired-end reads, resulting in a total of 199.10 million reads. Raw data in fastq format were initially processed using the fastp software (v0.21.0). In this step, clean data (clean reads) were obtained by removing reads containing adapters, reads containing poly-N, and low-quality reads from the raw data. Concurrently, the Q20, Q30, GC-content, and sequence duplication level of the clean data were calculated. The proportion of Q30 in the clean data was over 93% for each sample (Table 1).
Transcriptome data analysis
High-quality clean data was mapped to the Chinese soft-shell turtle reference genome (PelSin_1.0) using HISAT2 software (v2.2.1)12. Subsequently, the obtained BAM files were sorted using SAMtools (v1.2.1)13. Transcript assembly was then carried out using StringTie (v2.1.7)14. Finally, raw counts were obtained using the prepDE.py3 script from StringTie.
Differential gene expression analysis
To investigate potential regulatory differences between ICD and anti-ICD mechanisms for the interdigital tissues, we identified DEGs between the regressed (FW1-2 and HW1-2) and non-regressed (FW3-4 and HW3-4) webs of the turtles. DESeq215 software was employed to identify DEGs. Genes with a log2(Fold Change) ≥ 1 and adjusted P-value < 0.05 were defined as significantly differentially expressed genes (DEGs).
WISH
Partial coding sequence for Lhx9, serving as a probe, was amplified using a pair of specifically designed primers (forward primer: 5′-TGCTGGTTGTGGAGGGAAG-3′; reverse primer: 5′-GGCTGCTGGTCTCTGTCC-3′; product: 567 bp). Probes for Hoxd11 were generated using primers as described previously16. The WISH procedure adhered to previously published protocols17. Briefly, embryos were fixed in 4% PFA overnight, then dehydrated via a methanol gradient and stored at −80 °C. Before hybridization, they were rehydrated with a methanol gradient. For digestion, embryos were pre-treated with 0.2 M HCl and then incubated with 10 μg/ml proteinase K. Subsequently, embryos were washed in PBST and refixed in 0.2% glutaraldehyde within 4% PFA. They were then placed in a preheated hybridization solution with 1–2 μg/ml DIG-labeled probe and incubated at 65 °C overnight. After hybridization, the washed embryos were treated with blocking solution and incubated overnight with the anti-DIG antibody coupled with alkaline phosphatase. Antibodies were then removed, and the embryos were incubated in NTMT containing NBT-BCIP (Roche) until the desired color was achieved, at which point the reaction was stopped. To minimize background, embryos were washed at least six times overnight in 1% Triton X-100 solution. Finally, embryos were fixed in 4% PFA and photographed with a Leica microscope (M165FC).
RT-qPCR
Reactions were carried out on a LightCycler® 96 real-time PCR detection system (Roche), following the manufacturer’s guidelines. To determine relative mRNA levels, the expressions of target genes were normalized against the endogenous reference gene GAPDH. The 2–∆∆Ct method was used for calculations18. Three replicates were performed.
Data Records
Raw RNA-seq data files and TPM values are available on the Gene Expression Omnibus (GEO) database with accession no. GSE28705519. The data of DEGs are available from the figshare repository (https://doi.org/10.6084/m9.figshare.28255607)20.
Technical Validation
Hierarchical clustering analysis (HCA)
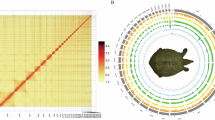
To verify the accuracy of sample collection and sequencing, we evaluated the similarity among samples. Firstly, a raw count expression matrix of all 19,328 genes was normalized using the TMM (Trimmed Mean of M-values) normalization method21. Spearman correlation coefficients were then calculated for the normalized matrix, and a hierarchical clustering analysis was performed using R (version 4.3.1). The results were visualized in the form of a heatmap that showed samples from the corresponding parts of the fore- and hindlimbs clustered together, with W1-2 separating from W3-4, samples that respectively represent the apoptotic webbed foot and the retained webbed foot (Fig. 2a). These results were consistent with the biological process, and thus effectively verify the accuracy and reliability of the sequence data.

HCA, PCA, and GO enrichment analysis. (a) Heatmap of the Spearman’s rank correlation coefficients and cluster analysis using the TMM normalized counts. R, Spearman’s rank correlation coefficients. (b) 3D PCA of samples based on the TPM counts. (c) Top 15 terms from the GO enrichment analysis of the DEGs, which identify three pathways known to be directly related to ICD that are highlighted in red.
Principal component analysis (PCA)
Three-dimensional (3D) PCA based on the TPM data shows that PC1 distinguishes the positional order from W1 to W4 and separates the fore- and hindlimb samples of W1 and W2 from those of W3 and W4 into two distinct clusters. These clusters are consistent with the HCA results, where the ICD samples cluster together and the anti-ICD samples cluster together. PC3 clearly distinguishes the fore- and hindlimb samples (Fig. 2b). In summary, the clustering by PCA supports the accuracy of the sequencing data.
Enrichment analysis
Gene Ontology (GO) analysis was performed on the 608 DEGs using the Database for Annotation, Visualization, and Integrated Discovery (DAVID, 2024q2 release, https://david.ncifcrf.gov/), with a significance threshold set at P < 0.05. The retinoic acid catabolic process, cell-cell adhesion mediated by cadherin, and BMP signaling pathway, regulatory pathways that have been reported to be directly related to ICD22, were all overrepresented (Fig. 2c). These results indicate that our data and screening methods are reliable.
Experimental Verification of the RNA-seq data
To validate the RNA-seq data, six genes were selected, 3 upregulated and 3 downregulated, from the top differentially expressed gene sets. This validation set including four conserved transcription factor genes associated with limb development (Runx3, Lhx9, Hoxd10 and Hoxd11) and two genes involved in epithelial cells or skin development (Msln and Phex). For validation using whole embryo in situ hybridization (WISH), genes for 2 transcription factors with higher expression levels, Lhx9 and Hoxd11, were chosen. The remaining four genes were used for verification by RT-qPCR. The heatmap of the RNA-seq data shows that there is a significant difference between the ICD and anti-ICD groups, with the patterns being consistent between the fore- and hindlimbs (Fig. 3a). The WISH results for Lhx9 and Hoxd11 in the turtle web are spatially localized in their expression patterns (Fig. 3b), which are consistent with the RNA-seq heatmap results, confirming the reliability of the data classification. Statistical analysis of the RT-qPCR results was performed using the SPSS software package (IBM SPSS Statistics for Windows, version 29.0). The RT-qPCR results show that the expression patterns for Runx3, Msln, Phex and Hoxd10 were consistent with the RNA-seq data (Fig. 3c). In conclusion, our validation experiments confirm the precision of the transcriptome data.

The RNA-seq data distribution patterns of six representative genes and their experimental validation. (a) Heatmap clustering of the TMM data for six genes. (b) WISH experimental validation for Lhx9 and Hoxd11 in both the fore- and hindlimbs is consistent with the RNA-seq data, showing that Lhx9 is highly expressed in regions undergoing ICD, while Hoxd11 is highly expressed in regions that are anti-ICD. Dorsal view of the autopod is shown with the anterior of the embryo up and the distal side to the right. Bar = 0.5 mm. (c) Gene expression determined by RT-qPCR. Data are shown as the mean ± SD. Different letters above the columns indicate significant differences among the samples (Duncan’s multiple test, P < 0.05).
Code availability
No custom code was created or used. All analytical methods and software are provided in the Methods.
References
Fallon, J. F. & Cameron, J. Interdigital cell death during limb development of the turtle and lizard with an interpretation of evolutionary significance. Development 40, 285–289, https://doi.org/10.1242/dev.40.1.285 (1977).
Weatherbee, S. D., Behringer, R. R., Rasweiler, J. J. IV & Niswander, L. A. Interdigital webbing retention in bat wings illustrates genetic changes underlying amniote limb diversification. Pro Natl Acad Sci USA 103, 15103–15107, https://doi.org/10.1073/pnas.0604934103 (2006).
Cooper, L. N. et al. Review and experimental evaluation of the embryonic development and evolutionary history of flipper development and hyperphalangy in dolphins (Cetacea: Mammalia). Genesis 56, e23076, https://doi.org/10.1002/dvg.23076 (2018).
Tokita, M., Matsushita, H. & Asakura, Y. Developmental mechanisms underlying webbed foot morphological diversity in waterbirds. Sci Rep 10, 8028, https://doi.org/10.1038/s41598-020-64786-8 (2020).
Cordeiro, I. R., Yu, R. & Tanaka, M. Regulation of the limb shape during the development of the Chinese softshell turtles. Evol Dev 22, 451–462, https://doi.org/10.1111/ede.12352 (2020).
Díaz-Hernández, M. E., Rios-Flores, A. J., Abarca-Buis, R. F., Bustamante, M. & Chimal-Monroy, J. Molecular control of interdigital cell death and cell differentiation by retinoic acid during digit development. J Dev Biol 2, 138–157, https://doi.org/10.3390/jdb2020138 (2014).
Kaltcheva, M. M., Anderson, M. J., Harfe, B. D. & Lewandoski, M. BMPs are direct triggers of interdigital programmed cell death. Dev Biol 411, 266–276, https://doi.org/10.1016/j.ydbio.2015.12.016 (2016).
Zuzarte-Luis, V., Berciano, M., Lafarga, M. & Hurle, J. Caspase redundancy and release of mitochondrial apoptotic factors characterize interdigital apoptosis. Apoptosis 11, 701–715, https://doi.org/10.1007/s10495-006-5481-8 (2006).
Díaz-Mendoza, M. J., Lorda-Diez, C. I., Montero, J., Garcia-Porrero, J. & Hurle, J. Interdigital cell death in the embryonic limb is associated with depletion of Reelin in the extracellular matrix. Cell Death Dis 4, e800–e800, https://doi.org/10.1038/cddis.2013.322 (2013).
Wang, Z. et al. The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan. Nat Genet 45, 701–706, https://doi.org/10.1038/ng.2615 (2013).
Tokita, M. & Kuratani, S. Normal embryonic stages of the Chinese softshelled turtle Pelodiscus sinensis (Trionychidae). Zoolog Sci 18, 705–715, https://doi.org/10.2108/zsj.18.705 (2001).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11, 1650–1667, https://doi.org/10.1038/nprot.2016.095 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq 2. Genome Biol 15, 1–21, https://doi.org/10.1186/s13059-014-0550-8 (2014).
De Bakker, M. A. et al. Selection on phalanx development in the evolution of the bird wing. Mol Biol and Evol 38, 4222–4237, https://doi.org/10.1093/molbev/msab150 (2021).
Hargrave, M., Bowles, J. & Koopman, P. In situ hybridization of whole-mount embryos. Methods Mol Biol, 103–113, https://doi.org/10.1385/1-59745-007-3:103 (2006).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 25, 402–408, https://doi.org/10.1006/meth.2001.1262 (2001).
Bai, S., Li, S. & Wang, Z. Transcriptome data of the interdigital webbing in Chinese Soft-shell Turtle (Pelodiscus sinensis). NCBI Gene Expression Omnibus (GEO) https://identifiers.org/geo/GSE287055 (2025).
Bai, S., Li, S. & Wang, Z. Transcriptome data from the interdigital webs of the Chinese Soft-shell Turtle (Pelodiscus sinensis). Figshare https://doi.org/10.6084/m9.figshare.28255607 (2025).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11, 1–9, https://doi.org/10.1186/gb-2010-11-3-r25 (2010).
Montero, J. A., Lorda-Diez, C. I. & Hurle, J. M. Confluence of cellular degradation pathways during interdigital tissue remodeling in embryonic tetrapods. Front Cell Dev Biol 8, 593761, https://doi.org/10.3389/fcell.2020.593761 (2020).
Acknowledgements
This work was supported by grants from the Department of Science & Technology of Liaoning Province (no. 2024-MS-099) and the Organization Department of Liaoning Provincial Committee of China (Liaoning Revitalization Talents Program, no. XLYC1907018).
Author information
Authors and Affiliations
Contributions
Z.W., S.B., S.L. and S.Z. designed the study; S.B., S.L., Y.T. and S.S. collected the samples; S.B. performed bioinformatics analyses; Y.T. and Z.J. verified the data; S.B. and S.L. drafted the manuscript; Z.W. and D.M.I. revised the manuscript; Z.W. and S.Z. contributed to funding acquisition. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bai, S., Li, S., Tang, Y. et al. Transcriptome data from the interdigital webs of the Chinese Soft-shell Turtle (Pelodiscus sinensis). Sci Data 12, 806 (2025). https://doi.org/10.1038/s41597-025-05188-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05188-1
