
Abstract
Framing the investigation of diverse cancers as a machine learning problem has recently shown significant potential in multi-omics analysis and cancer research. Empowering these successful machine learning models are the high-quality training datasets with sufficient data volume and adequate preprocessing. However, while there exist several public data portals, including The Cancer Genome Atlas (TCGA) multi-omics initiative or open-bases such as the LinkedOmics, these databases are not off-the-shelf for existing machine learning models. In this paper, we introduce MLOmics, an open cancer multi-omics database aiming at serving better the development and evaluation of bioinformatics and machine learning models. MLOmics contains 8,314 patient samples covering all 32 cancer types with four omics types, stratified features, and extensive baselines. Complementary support for downstream analysis and bio-knowledge linking are also included to support interdisciplinary analysis.
Similar content being viewed by others
Background & Summary
Multi-omics analysis has shown great potential to accelerate cancer research. A promising trend consists of framing the investigation of diverse cancers as a machine learning problem, where complex molecular interactions and dysregulations associated with specific tumor cohorts are revealed through integration of multi-omics data into machine learning models. Several impressive achievements have been demonstrated in molecular subtyping1,2,3, disease-gene association prediction4,5,6, and drug discovery7.
Empowering successful machine learning models are the high-quality training datasets with sufficient data volume and adequate preprocessing. While there exist several public data portals including The Cancer Genome Atlas (TCGA) multi-omics initiative8 or open-bases such as the LinkedOmics9, these databases are not off-the-shelf for existing machine learning models. To make these data model-ready, a series of laborious, task-specific processing steps such as metadata review, sample linking, and data cleaning are mandatory. The domain knowledge required, as well as a deep understanding of diverse medical data types and proficiency in bioinformatics tools have become an obstacle for researchers outside of such backgrounds. The gap between the growing body of powerful machine learning models and the absence of well-prepared public data has become a major bottleneck. Currently, some existing methods validate their proposed machine learning models using inconsistent experimental protocols, with variations in datasets, data processing techniques and evaluation strategies10. These studies could benefit from a fair assessment of extensive baselines on a uniform footing with unified datasets and to offer more reliable recommendations on models.
To meet the growing demand of the community, we introduce MLOmics, an open cancer multi-omics database aiming at serving better the development and evaluation of bioinformatics and machine learning models. MLOmics collected 8,314 patient samples covering all 32 cancer types from the TCGA project. All samples were uniformly processed to contain four omics types: mRNA expression, microRNA expression, DNA methylation, and copy number variations, followed by categorization, protocol verification, feature profiling, transformation, and annotation. For each dataset, we provide three feature versions: Original, Aligned, and Top, to support feasible analysis. For example, the Top version contains the most significant features selected via the ANOVA test11 across all samples to filter out potentially noisy genes. The MLOmics datasets were carefully examined with 6 ~ 10 highly cited baseline methods. These baselines were rigorously reproduced and evaluated with various metrics to ensure fair comparison. Complementary resources are included to support basic downstream biological analysis, such as clustering visualization, survival analysis, and Volcano plots. Last but not least, we provide support to interdisciplinary analysis via our locally deployed bio-base resources. Interdisciplinary researchers can retrieve and integrate bio-knowledge of cancer omics studies through resources such as the STRING12 and KEGG13. For instance, exploration of bio-network inference14 and simulated gene knockouts15 is supported. In summary, MLOmics is an open, unified database approachable to non-experts for developing/evaluating machine learning models; conducting interdisciplinary analysis and supporting cancer research and broader biological studies. We showcase the above usages in the Usage Notes section. A detailed overview of the MLOmics database and its characteristics is provided in Fig. 1.

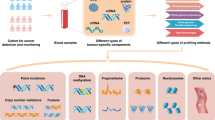
Schematic workflow of creating the MLOmics. The process starts with collecting patient samples covering 32 cancer types from the TCGA project. All resources in diverse data types and sizes are uniformly integrated and processed to contain data of four omics types. Datasets for benchmark ML tasks were constructed based on the processed data. MLOmics also selected baselines, metrics, and resources to support downstream biological analysis. Overview of the MLOmics. MLOmics provides an interface for developing and evaluating machine learning models based on cancer multi-omics data. MLOmics provides datasets in three feature scales for 20 classification, clustering, and omics imputation learning tasks. MLOmics also provides statistical, ML, and DL baselines for each task, which are evaluated by fair metrics. Bio-knowledge database linking with MLOmics. MLOmics provides resources to link with other bio-knowledge databases, enabling the integration of outer resources for applications such as ML evaluation, gene-disease association exploration, network inference, and functional analysis.
Methods
Data Collection and Preprocessing
The data were sourced from TCGA via the Genomic Data Commons (GDC) Data Portal16. The original resources in TCGA are organized by cancer type, and the omics data for individual patients are scattered across multiple repositories. Therefore, retrieving and collecting omics data require linking samples with metadata and applying different preprocessing protocols. MLOmics employs a unified pipeline that integrates data preprocessing, quality control, and multi-omics assembly for each patient, followed by alignment with their respective cancer types. Specifically, we perform different steps for each omics type as follows:
Transcriptomics (mRNA and miRNA) data: 1. Identifying transcriptomics. We trace the downloaded data using the “experimental_strategy” field in the metadata, marked as “mRNA-Seq” or “miRNA-Seq”, then we verify that “data_category” is labeled as “Transcriptome Profiling.” 2. Determining experimental platform. We identify the experimental platform from the metadata, such as “platform: Illumina” or “workflow_type: BCGSC miRNA Profiling.” 3. Converting gene-level estimates. For data generated by platforms like Illumina Hi-Seq, use the edgeR package17 to convert scaled gene-level RSEM estimates into FPKM values. 4. Non-Human miRNA filtering. For “miRNA-Seq” data from platforms like Illumina GA and Agilent arrays, we identify and remove non-human miRNA expressions using species annotations from databases such as miRBase18. 5. Noise eliminating. We remove features with zero expression in more than 10% of samples or those with undefined value (N/A). 6. Transformation. Finally, we apply logarithmic transformations to obtain the log-converted mRNA and miRNA data.
Genomic (CNV) data: 1. Identifying CNV Alterations. We examine how gene copy-number alterations are recorded in the metadata, using key descriptions such as “Calls made after normal contamination correction and CNV removal using thresholds.” 2. Filter Somatic Mutations. We capture only somatic variants by retaining entries marked as “somatic” and filtering out germline mutations. 3. Identify Recurrent Alterations. We use the GAIA package19 to identify recurrent genomic alterations in the cancer genome, based on raw data representing all aberrant regions from copy-number variation segmentation. 4. Annotate Genomic Regions. We annotate the recurrent aberrant genomic regions using the BiomaRt package20.
Epigenomic (Methy) data: 1. Identify Methylation Regions. We examine how methylation is defined in metadata to map methylation regions to genes, using key descriptions like “Average methylation (beta-values) of promoters defined as 500bp upstream & 50 downstream of Transcription Start Site (TSS)” or “With coverage >= 20 in 70% of the tumor samples” 2. Normalize Methylation Data. We perform median-centering normalization to adjust for systematic biases and technical variations across samples, using the R package limma21. 3. Select Promoters with Minimum Methylation. For genes with multiple promoters, we select the promoter with the lowest methylation levels in normal tissues.
After processing the omics sources, the data are annotated with unified gene IDs to resolve variations in naming conventions caused by the difference in sequencing methods or reference standards22. Then, the omics data are aligned across multiple sources based on their corresponding sample IDs. Finally, the data files are organized by cancer type for further dataset construction.
Datasets Construction
MLOmics reorganizes the collected and processed data resources into different feature versions tailored to various machine learning tasks. For each task, MLOmics provides several baselines, evaluation metrics, and the ability to link with biological databases such as STRING and KEGG for further biological analysis of different machine learning models.
Feature Processing
Machine learning models require tabular data with a the same number of features across samples. In addition to the Original feature scale that contains a full set of genes (variations included) directly extracted from the collected omics files, MLOmics provides two other well-processed feature scales: Aligned and Top. The former scale filters out non-overlapping genes and selects the genes shared across different cancer types; and the latter identifies the most significant features. Specifically, the following steps are performed for each scale:
Aligned: 1. we resolve the mismatches in gene naming formats such as ensuring compatibility between cancers that use different reference genomes. 2. we identify the intersection of feature lists across datasets to ensure all selected features are present in different cancers. 3. we conduct z-score feature normalization.
Top: 1. we perform multi-class ANOVA11 to identify genes with significant variance across multiple cancer types. 2. we perform multiple testing using the Benjamini-Hochberg (BH)23 correction to control the false discovery rate (FDR)24. 3. the features are ranked by the adjusted p-values p < 0.05 or by the user-specified scales). 4. we conduct z-score feature normalization which reduces the presence of non-significant genes across cancers and this could be beneficial for biomarker studies.
20 Task-ready Datasets with Baselines and Metrics
We provide 20 off-the-shelf datasets ready for machine learning models ranging from pan-cancer/cancer subtype classification, subtype clustering to omics data imputation. We also include well recognized baselines that leveraged classical statistical approaches and machine/deep learning methods as well as metrics for standard evaluation.
Pan-cancer and golden-standard cancer subtype classification. Pan-cancer classification aims to identify each patient’s specific cancer type. Moreover, a cancer typically comprises multiple subtypes that differ in their biochemical profiles. Some subtypes have been well-studied in prior research and widely accepted as the golden standard. We re-label patient samples to support subtyping evaluation. These two classification tasks potentially improve cancer early diagnostic accuracy and treatment outcomes.
Datasets: MLOmics provides six labeled datasets: one pan-cancer dataset and five gold-standard subtype datasets (GS-COAD, GS-BRCA, GS-GBM, GS-LGG, and GS-OV).
Baselines: we opt for the following classical classification methods as baselines: XGBoost25, Support Vector Machines (SVM)26, Random Forest (RF)27, and Logistic Regression (LR)28. Additionally, we include six popular, open-sourced deep learning methods: Subtype-GAN29, DCAP30, XOmiVAE31, CustOmics32, and DeepCC33.
Metrics: For classification evaluation, we opt for precision (Pre), recall (Re), and F1-score (F1). Since clustering is the primary focus of the subtyping task, due to the limited sample size (typically <100), we provide normalized mutual information (NMI) and adjusted rand index (ARI) to evaluate the agreement between the clustering results obtained by different methods and the true labels.
Cancer Subtype Clustering. Cancer subtyping remains an open question under fierce debate for most cancers, especially rare types. Numerous studies propose various clustering methods to identify distinct groups by identifying different clusters to support downstream evaluation and discovery of new subtypes.
Datasets: MLOmics provides nine unlabeled rare cancer datasets (ACC, KIRP, KIRC, LIHC, LUAD, LUSC, PRAD, THCA, and THYM) for this learning task.
Baselines: In addition to the aforementioned Subtype-GAN, DCAP, MAUI, XOmiVAE, we also include six clustering methods: Similarity Network Fusion (SNF)34, Neighborhood-based Multi-Omics clustering (NEMO)35, Cancer Integration via Multi-kernel Learning (CIMLR)36, iClusterBayes37, moCluster38, and MCluster-VAEs39.
Metrics: To evaluate the goodness of clustering we opt for the classic Silhouette coefficient (SIL)40 and log-rank test p-value on survival time (LPS)41.
Omics Data Imputation. In addition to classification and clustering, we also provide a data imputation task focusing on imputing multi-omics cancer data. The collected omics data typically have missing values due to experimental limitations, technical errors, or inherent variability. The imputation process is crucial for ensuring the integrity and usability of TCGA omics data42.
Datasets: MLOmics provides five omics datasets with missing values (Imp-BRCA, Imp-COAD, Imp-GBM, Imp-LGG, and Imp-OV). Given a full dataset as a matrix \(D\in {{\mathbb{R}}}^{n\times m}\), we follow prior works42,43 to generate a mask matrix M ∈ {0, 1}n×m uniformly at random with the probability of missing P(Mij = 0) = rmiss, and the probability of retaining P(Mij = 1) = 1 − rmiss. The final data matrix is obtained by multiplying element-wise the data matrix D with the mask M. The missing level rmiss is selected from [0.3, 0.5, 0.7].
Baselines: We opt for seven well-recognized methods for imputing missing values, including: Mean Imputation that imputes the missing entry with mean values of entries around it (Mean); K-Nearest Neighbors (KNN) that imputes the missing value with the weighted Euclidean K nearest neighbors; Multivariate Imputation by Chained Equations (MICE) that performs multiple regression to model each missing value conditioned on non-missing values44; Iterative SVD (iSVD) that imputes by iterative low-rank SVD decomposition45; Spectral Regularization Algorithm (Spectral) that also employs SVD but with a soft threshold and the nuclear norm regularization46; Generative Adversarial Imputation Nets (GAIN) that proposes to distinguish between fake and true missing patterns by the generator-discriminator architecture43; Graph Neural Network for Tabular Data (GRAPE) that utilizes the graph networks to impute based on learned information from columns and rows of the data matrix42.
Metrics: We use the Mean Squared Error (MSE) between the unmasked entries (Mij = 1) as the training loss to let the model predict the actual value. During the test, the masked missing values are used for evaluating the model performance (Mij = 0).
MLOmics will be continuously updated with baselines and evaluations on the defined learning tasks. A detailed description of the baselines and metrics is provided in the Supplementary Material.
Data Records
The MLOmics main datasets47 are available on Figshare (https://figshare.com/articles/dataset/MLOmics_Cancer_Multi-Omics_Database_for_Machine_Learning/28729127) and Hugging Face (https://huggingface.co/datasets/AIBIC/MLOmics). The MLOmic main datasets are now accessible under the Creative Commons 4.0 Attribution (CC-BY-4.0), which supports its use for educational and research purposes. The main datasets presented include all cancer multi-omics datasets corresponding to the various tasks described above.
MLOmics Overview
The MLOmics repository is structured into three sections: (1) Main Datasets, (2) Baseline and Metrics, and (3) Downstream Analysis Tools and Resources Linking.
The core section is the (1) Main Datasets: the repository hosts a comprehensive collection of tasks-ready cancer multi-omics datasets, stored primarily as .csv (comma-separated values) files.
Along with the other two sections: (2) Baseline and Metrics: the repository provides source codes of baseline models and evaluation metrics for different tasks, typically implemented in Python or R code; (3) Downstream Analysis Tools and Resources Linking: the repository encompasses additional tools and resources that complement the main datasets and downstream omics analysis needs, implemented as .csv, .py or R files.
In the following parts of this section, we focus on presenting the properties of the Main Datasets, detailing its organizational structure, principal components, and resources.
Main Datasets Format
Main datasets of MLOmics use .csv files to manage and store all omics datasets, widely favored in biomedical research, including multi-omics studies. Despite its simplicity, .csv remains efficient even with large datasets encountered in genomic and proteomic studies. Both Python and R provide built-in functions and libraries to read, write, and analyze .csv files efficiently.
Specifically, MLOmics provides its main datasets in .csv files as plain-text files, where commas separate data values. These files maintain a structured and intuitive format, with rows representing individual data records and columns corresponding to different attributes or variables. The key characteristics of the main dataset files are:
-
Encoding: UTF-8, ensuring broad compatibility across different systems and software.
-
Delimiter:, (comma), used to separate values in each row.
-
Line Ending: LF (\n) for Unix/Linux or CRLF (\r\n) for Windows, ensuring proper formatting across operating systems.
-
Header Row: The first row contains column names, typically representing sample identifiers (e.g., Sample1, Sample2, Sample3, etc.).
-
Data Rows: Each subsequent row corresponds to a distinct feature, such as a gene, with attributes like expression values recorded across multiple patient samples.
-
Values: Numeric values (float), representing continuous measurements of omics attributes.
For example, consider a simplified .csv file containing mRNA data for a set of gene features (rows) across several patient samples (columns):
Feature | Sample1 | Sample2 | Sample3 | Sample4 |
GeneA | 0.23 | 0.18 | 0.35 | 0.21 |
GeneB | 0.56 | 0.49 | 0.52 | 0.58 |
GeneC | 0.19 | 0.22 | 0.15 | 0.17 |
GeneD | 0.08 | 0.10 | 0.09 | 0.12 |
In this example, each row corresponds to a specific gene (GeneA, GeneB, GeneC, GeneD). Each column represents a different sample (Sample1, Sample2, Sample3, Sample4). The numeric values (variables) in the cells denote the expression levels of each gene in each sample.
Main Datasets Structure
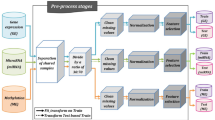
As shown in Fig. 2, the main datasets repository is organized into three layers:

Schematic of MLOmics resources structure. The MLOmics framework consists of three major components. The Main Datasets file includes all cancer multi-omics datasets for various tasks. The Baseline and Metrics file provides the source code for baseline models and performance metrics. The Downstream Analysis Tools and Resources Linking file offers sources for further analysis and links to additional biological resources.
(1) The first layer contains three files corresponding to three different tasks: Classification_datasets, Clustering_datasets, and Imputation_datasets.
(2) The second layer includes files for specific tasks, such as GS-BRCA, ACC, and Imp-BRCA.
(3) The third layer contains three files corresponding to different feature scales, i.e., Original, Aligned, and Top.
The omics data from different omics sources are stored in the following files: Cancer_miRNA_Feature.csv, Cancer_mRNA_Feature.csv, Cancer_CNV_Feature.csv, and Cancer_Methy_Feature.csv. Here, Cancer represents the cancer type, and Feature indicates the feature scale type.
The ground truth labels are provided in the file Cancer_label_num.csv, where Cancer represents the cancer type. The patient survival records are stored in the file Cancer_survival.csv.
Technical Validation
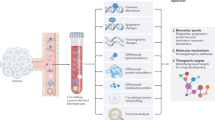
The Methods section detailed the data collection, preprocessing and curation of the MLOmics datasets. To validate the collected datasets, we set up a series of classification, clustering and imputation experiments each with a wide array of models ranging from conventional statistical models to deep neural networks. The experimental results and evaluations are summarized in Fig. 3.

Experimental results and downstream analyses of machine learning baselines applied to MLOmics datasets. (a) PREC bar plots for each baseline method across all datasets. Overall, machine learning-based methods (Subtype-GAN, DCAP, MAUI, XOmiVAE, CustOmics, and DeepCC) outperformed traditional statistical methods (SVM, XGBoost, RF, and LR). (b) SIL heatmaps for each baseline method across all datasets. Methods employing deep generative neural network architectures (Subtype-GAN, DCAP, MAUI, XOmiVAE, and MCluster-VAEs) generally outperformed other methods (SNF, NEMO, CIMLR, iClusterBayes, and moCluster). (c) Box plots for each baseline method across three imputation datasets. Matrix decomposition methods (SVD, Spectral) outperformed deep learning-based methods (GAIN, GNN). (d,e) Schematic illustrations of downstream analysis results based on the clustering outcomes of XOmiVAE applied to specific cancer patient clustering datasets. In the survival analysis plot, survival curves in different colors correspond to distinct clustering groups. In the volcano plot and KEGG pathway analysis, red and blue indicate downregulated and upregulated genes between patient groups, respectively. In the patient group clustering plot, different colors represent samples belonging to different clusters. In the simulated gene knockout analysis, red and green dots indicate sample clustering before and after expression knockout, respectively.
Usage Notes
We provide comprehensive guidelines for utilizing the MLOmics repository, as illustrated in Fig. 4. All codes and files are ready for direct loading and analysis using standard Python data packages such as NumPy and Pandas.

The code usage note of MLOmics. The MLOmics can be used following the illustrated code usage instructions, including MLOmics setup, code usage for custom model-task-dataset specifications, and code for downstream analysis workflows.
As a demonstration of how to use MLOmics for machine learning applications, we provide 20 classification, clustering, and imputation tasks with fair evaluation protocols for pan-cancer analysis, cancer subtypes, and omics imputation. Access to these resources is provided in the Code Availability section. A rising trend in multi-omics analysis is to integrate multi-omics data (non-network data) with biological networks to better understand complex functions on the gene or protein level. MLOmics provides offline linking resources for well-established databases such as STRING12 and KEGG13. We hope it can lower the barriers to entry for machine learning researchers interested in developing methods for cancer multi-omics data analysis, thereby encouraging rapid progress in the field.
Code availability
All codes and resources of MLOmics are publicly available under the CC-BY-4.0. The code for preprocessing and preparing the main MLOmics databases, as well as the benchmarking algorithms used in MLOmics, is available at the following repository: (https://github.com/chenzRG/Cancer-Multi-Omics-Benchmark).
References
Picard, M., Scott-Boyer, M.-P., Bodein, A., Périn, O. & Droit, A. Integration strategies of multi-omics data for machine learning analysis. Comput. Struct. Biotechnol. J. 19, 3735–3746 (2021).
Withnell, E., Zhang, X., Sun, K. & Guo, Y. Xomivae: an interpretable deep learning model for cancer classification using high-dimensional omics data. Briefings Bioinforma 22 (2021).
Chen, Z., Zhu, L., Yang, Z. & Matsubara, T. Automated cancer subtyping via vector quantization mutual information maximization. In Joint European Conference on Machine Learning and Knowledge Discovery in Databases, 88–103 (Springer, 2022).
Gysi, D. M., Voigt, A., Fragoso, T.d. M., Almaas, E. & Nowick, K. wto: an r package for computing weighted topological overlap and a consensus network with integrated visualization tool. BMC bioinformatics 19, 1–16 (2018).
Wu, Q.-W., Xia, J.-F., Ni, J.-C. & Zheng, C.-H. Gaerf: predicting lncrna-disease associations by graph auto-encoder and random forest. Briefings Bioinforma. 22, bbaa391 (2021).
Sharma, D. & Xu, W. ReGeNNe: genetic pathway-based deep neural network using canonical correlation regularizer for disease prediction. Bioinformatics 39, btad679, https://doi.org/10.1093/bioinformatics/btad679 (2023).
Vamathevan, J. et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 18, 1, https://doi.org/10.1038/s41573-019-0024-5 (2019).
The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49, https://doi.org/10.1038/nature12222 (2013).
Vasaikar, S., Straub, P., Wang, J. & Zhang, B. Linkedomics: Analyzing multi-omics data within and across 32 cancer types. Nucleic acids research 46 (2017).
Reel, P. S., Reel, S., Pearson, E., Trucco, E. & Jefferson, E. Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. advances 49, 107739 (2021).
St, L. & Wold, S. et al. Analysis of variance (anova). Chemom. intelligent laboratory systems 6, 259–272 (1989).
Szklarczyk, D. et al. The string database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic acids research 51, D638–D646 (2023).
Kanehisa, M. & Goto, S. Kegg: kyoto encyclopedia of genes and genomes. Nucleic acids research 28, 27–30 (2000).
Zitnik, M. et al. Current and future directions in network biology. Bioinforma. Adv. 4, vbae099 (2024).
Bergman, A. & Siegal, M. L. Evolutionary capacitance as a general feature of complex gene networks. Nature 424, 549–552 (2003).
Grossman, R. L. et al. Toward a shared vision for cancer genomic data. New Engl. J. Medicine 375, 1109–1112 (2016).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edger: a bioconductor package for differential expression analysis of digital gene expression data. bioinformatics 26, 139–140 (2010).
Kozomara, A., Birgaoanu, M. & Griffiths-Jones, S. mirbase: from microrna sequences to function. Nucleic acids research 47, D155–D162 (2019).
al. SMe. gaia: GAIA: An R package for genomic analysis of significant chromosomal aberrations R package version 2.39.0 (2021).
Durinck, S. et al. Biomart and bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440 (2005).
Ritchie, M. E. et al. limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic acids research 43, e47–e47 (2015).
Tomczak, K., Czerwińska, P. & Wiznerowicz, M. Review the cancer genome atlas (tcga): an immeasurable source of knowledge. Contemp. Oncol. Onkologia 2015, 68–77 (2015).
Thissen, D., Steinberg, L. & Kuang, D. Quick and easy implementation of the benjamini-hochberg procedure for controlling the false positive rate in multiple comparisons. J. educational behavioral statistics 27, 77–83 (2002).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Royal statistical society: series B (Methodological) 57, 289–300 (1995).
Chen, T. & Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd acm sigkdd international conference on knowledge discovery and data mining, 785–794 (2016).
Burges, C. J. A tutorial on support vector machines for pattern recognition. Data mining and knowledge discovery 2, 121–167 (1998).
Breiman, L. Random forests. Mach. learning 45, 5–32 (2001).
Sperandei, S. Understanding logistic regression analysis. Biochem. medica 24, 12–18 (2014).
Yang, H., Chen, R., Li, D. & Wang, Z. Subtype-gan: a deep learning approach for integrative cancer subtyping of multi-omics data. Bioinformatics 37, 2231–2237 (2021).
Chai, H. et al. Integrating multi-omics data through deep learning for accurate cancer prognosis prediction. Comput. biology medicine 134, 104481 (2021).
Withnell, E., Zhang, X., Sun, K. & Guo, Y. Xomivae: an interpretable deep learning model for cancer classification using high-dimensional omics data. Briefings Bioinforma. 22, bbab315 (2021).
Benkirane, H., Pradat, Y., Michiels, S. & Cournède, P.-H. Customics: A versatile deep-learning based strategy for multi-omics integration. PLoS Comput. Biol. 19, e1010921 (2023).
Gao, F. et al. Deepcc: a novel deep learning-based framework for cancer molecular subtype classification. Oncogenesis 8, 44 (2019).
Wang, B. et al. Similarity network fusion for aggregating data types on a genomic scale. Nat. methods 11, 333–337 (2014).
Rappoport, N. & Shamir, R. Nemo: cancer subtyping by integration of partial multi-omic data. Bioinformatics 35, 3348–3356 (2019).
Wilson, C. M., Li, K., Yu, X., Kuan, P.-F. & Wang, X. Multiple-kernel learning for genomic data mining and prediction. BMC bioinformatics 20, 1–7 (2019).
Mo, Q. et al. A fully bayesian latent variable model for integrative clustering analysis of multi-type omics data. Biostatistics 19, 71–86 (2018).
Meng, C., Helm, D., Frejno, M. & Kuster, B. mocluster: identifying joint patterns across multiple omics data sets. J. proteome research 15, 755–765 (2016).
Rong, Z. et al. Mcluster-vaes: an end-to-end variational deep learning-based clustering method for subtype discovery using multi-omics data. Comput. Biol. Medicine 150, 106085 (2022).
Shahapure, K. R. & Nicholas, C. Cluster quality analysis using silhouette score. In 2020 IEEE 7th international conference on data science and advanced analytics (DSAA), 747–748 (IEEE, 2020).
Xie, J. & Liu, C. Adjusted kaplan–meier estimator and log-rank test with inverse probability of treatment weighting for survival data. Stat. medicine 24, 3089–3110 (2005).
You, J., Ma, X., Ding, Y., Kochenderfer, M. J. & Leskovec, J. Handling missing data with graph representation learning. Adv. Neural Inf. Process. Syst. 33, 19075–19087 (2020).
Yoon, J., Jordon, J. & van der Schaar, M. GAIN: Missing data imputation using generative adversarial nets. In Proceedings of the 35th International Conference on Machine Learning, 5689–5698 (2018).
van Buuren, S. & Groothuis-Oudshoorn, K. mice: Multivariate imputation by chained equations in r. J. Stat. Softw. 1-67 (2011).
Troyanskaya, O. et al. Missing value estimation methods for dna microarrays. Bioinformatics 17, 520–525 (2001).
Mazumder, R., Hastie, T. & Tibshirani, R. Spectral regularization algorithms for learning large incomplete matrices. The J. Mach. Learn. Res. 11, 2287–2322 (2010).
Kotoge, R. Mlomics: Cancer multi-omics database for machine learning, https://doi.org/10.6084/m9.figshare.28729127.v1 (2025).
Acknowledgements
This work was supported by JSPS KAKENHI Grant-in-Aid for Scientific Research Number JP24K20778, JST CREST JPMJCR23M3, NSF award SCH-2205289, SCH-2014438, IIS-2034479.
Author information
Authors and Affiliations
Contributions
Zheng Chen and Ziwei Yang launched the project. Ziwei Yang collected the data and organized the datasets. Rikuto Kotoge built the dataset repository for storage and access. Ziwei Yang, Rikuto Kotoge, and Xihao Piao conducted the experiments. Zheng Chen, Lingwei Zhu, Ziwei Yang, and Peng Gao contributed to writing the manuscript and reviewed the article. Peng Gao contributed to the biological review. Zheng Chen, Yasuko Matsubara, Yasushi Sakurai, and Jimeng Sun conceptualized and supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, Z., Kotoge, R., Piao, X. et al. MLOmics: Cancer Multi-Omics Database for Machine Learning. Sci Data 12, 913 (2025). https://doi.org/10.1038/s41597-025-05235-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05235-x
