
Abstract
Sambus kanssuensis Ganglbauer, 1890 (Coleoptera: Buprestidae), distributed in Gansu and Sichuan Provinces of China, is a phytophagous pest that feeds on the toxic plant Buddleja. However, the genomic resources of this beetle remain unknown, which impedes the understanding of its ecological adaptations. Consequently, this study presents a complete, well-assembled, and annotated genome of S. kanssuensis. The assembled results indicate a genome size of 312.42 Mb, comprising 206 scaffolds, with an N50 of 34.04 Mb; 98.68% of the assembly sequences were anchored to 11 chromosomes, including one sex chromosome. The genome contains 12,723 protein-coding genes, of which 11,977 have been annotated. BUSCO analysis revealed that the completeness of the chromosome-level genome is 97.9%. This chromosome-level genome provides valuable data for further investigations into detoxification mechanisms, ecological adaptations, population genetics, and the evolution of Buprestidae.
Similar content being viewed by others

Background & Summary
During the long-term process of coevolution, a plant–insect arms race has developed between herbivorous insects and plants1. Plants can defend themselves chemically through secondary metabolites to prevent herbivory2,3,4,5,6, while insects have evolved corresponding counterstrategies6,7,8,9. Buddleja sp. is a toxic plant, whose sap can kill or paralyze fish and contains a variety of substances, including flavonoids and terpenoids. Species of the genus Sambus Deyrolle, 1864 feed on the flowers and leaves of Buddleja sp., exhibiting strong host plant specificity. This characteristic makes Sambus an ideal model for studying the adaptive mechanisms of insects in relation to their host plants.
The genus Sambus belongs to the subfamily Agrilinae of the family Buprestidae (order Coleopetera). This genus was transferred from Coraebini to Agrilini in 200010, however, its tribal status remains controversial11,12,13,14. It comprises approximately 150 known species worldwide, with the majority distributed in Southeast Asia and East Asia. Sambus kanssuensis Ganglbauer, 1890 (Fig. 1) is widely distributed in western Sichuan and southern Gansu of China. Its host is a toxic plant Buddleja sp.15,16,17,18,19,20. This buprestid species exhibits the significant sexual dimorphism: the frons is copper green in male, while purple bronze in female; and females are distinctly larger than males. The genome was sequenced using adults of S. kanssuensis collected from Kangding City of Sichuan Province, China.

The habitus of Sambus kanssuensis. (A) male, (B) female.
Currently, only genomes of four species have been sequenced and assembled in Buprestidae21,22, however, these genomes have not been annotated. In the present study, we sequenced, assembled and annotated the chromosome-level genome of S. kanssuensis. The complete genome size is 312.42 Mb, including 206 scaffolds, with an N50 of 34.04 Mb. A total of 12,723 protein-coding genes (PCGs) have been identified. The completeness of the genome assembly and annotation is 97.90% and 96.10%, respectively, based on Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis. This high-quality chromosomal-level genome of S. kanssuensis, described herein, will promote research on the taxonomy, ecology and evolution of the jewel beetles as well as the detoxification mechanisms of herbivorous insects.
Methods
Sample collection
In the present study, adult specimens of S. kanssuensis were collected from the plant Buddleja sp. at Paomashan Park (30.04288°N, 101.96951°E, elev. 2740 m) in Kangding City, Sichuan Province, China, on July 3, 2024. The collected specimens were temporarily stored in liquid nitrogen. After returning to the laboratory, the specimens were stored in an ultra-low temperature freezer at –85 °C. To prevent genetic contamination, the abdomen of the specimen is removed. Tissues from the head and thorax were used for genomic DNA extraction.
Genome sequencing
The total genomic DNA was extracted from 35 male adults using the cetyltrimethylammonium bromide (CTAB) method23. After removing impurities, the genomic DNA was sequenced using Illumina and PacBio technologies. The quality of the extracted DNA was assessed using 0.7% agarose gel electrophoresis, and the concentration of genomic DNA was quantified using a Qubit 3.0 fluorometer (Invitrogen, USA). For Pacbio HiFi sequencing, genomic DNA fragments underwent damage repair, adapter ligation, and fragment selection prior to the construction of the DNA library. The PCR-free Single Molecule Real Time (SMRT) bell library was constructed and sequenced using the PacBio Revio sequencing platform. Adapter sequences and low-quality reads were removed using High-Throughput Quality Control (HTQC) v1.92.31024 with default parameters. Data quality control and statistical analyses were performed using PacBio software SMRT Link v12.0 (–min-passes = 3 –min-rq = 0.99), resulting in the final valid data. Finally, a total of 17.67 Gb HiFi reads were obtained (total number: 949,468, average length: 18,607.6 bp, N50 length: 18,692 bp) and subsequently used for genome assembly. For short reads sequencing, the Nextera DNA Flex Library Prep Kit (Illumina, San Diego, USA) was used to construct an Illumina sequencing library with an insert size of 150 bp. High-throughput sequencing was performed using the Illumina NovaSeq6000 platform (Illumina, San Diego, USA). The raw reads were filtered, resulting in 21.63 Gb of clean data.
High throughput Chromosome Conformation Capture (Hi-C) technology was employed to facilitate chromosome-level genome assembly and to capture chromatin interactions throughout the entire genome. The DpnII enzyme was used to digest the purified cell nuclei. Following this, Hi-C samples were generated through a series of procedures, including end repair, biotin labelling, blunt-end ligation, DNA purification, and random shearing into fragments ranging from 300 to 700 bp. Sequencing libraries were prepared using the Plus DNA Library Prep Kit, with insert sizes ranging from 200 to 400 bp. After passing quality control, the libraries were sequenced on the Illumina NovaSeq6000 platform, generating 150 bp paired-end reads. Ultimately, we obtained 46.87 Gb of raw Hi-C reads for assembly.
For full-length transcriptome sequencing, the head tissue from 13 adult females was extracted using the Qiagen Kit (Qiagen Sciences, USA), following the manufacturer’s instructions. The SQK–PCS109 kit (Oxford Nanopore Technologies, UK) was used for the library construction. A specific concentration and volume of the cDNA library were then added to the flow cell, which was subsequently transferred to the Oxford Nanopore PromethION sequencer for real-time single-molecule sequencing. The data were filtered to remove sequences with an average quality score of less than or equal to 7. Finally, 12.96 Gb of valid data and 13,085,109 bp of total bases were obtained. The N50 and N90 of read length were 1,139 bp and 555 bp, respectively.
Survey of genome characteristics and genome assembly
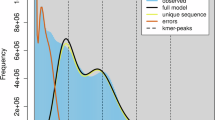
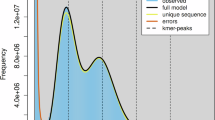
To assess the genome size and heterozygosity of S. kanssuensis, this study conducted a survey of genomic features using K-mer analysis25. The 19-mer frequency distribution analysis (Supplementary Table 1) was performed using Jellyfish v2.2.1026. Subsequently, the genome size and heterozygosity were estimated using GenomeScope v2.027. The predicted genome size of S. kanssuensis was 325.99 Mb, with a heterozygous ratio of 0.96, a duplication rate of 40.62%, and a GC content of 32.28% (Fig. 2).

The genome scope profile of Sambus kanssuensis.
The PacBio HiFi reads were converted to FASTA format using bam2fasta integrated in SAMtools v1.928. Genome assembly was performed using hifiasm v0.19.8-r60329 based on Overlap-Layout-Consensus (OLC) method. The primary assembly was polished with NextPolish v1.1.030. Purge_dups v1.2.531 was then applied to perform haplotype separation, resulting in the final draft genome. Minimap2 v2.17-r94132 was used for mapping reads during redundancy removal and short-read polishing steps. ALLHiC v0.9.833 (-e GATC) was employed to assist in the assembly of chromosomal-scale genomes. Two software tools, 3D-DNA v20100834 (-q 30) and Juicer v1.635 (-g matrial -s MboI -t 30 -S early), were used to anchor primary contigs into chromosomes. Juicebox v1.11.082636 (Coverage) was used to visualize Hi-C contact maps and manually correct errors. The completeness of the genome assembly was evaluated using BUSCO v5.4.737 (-evalue 1e-05) based on the database insecta_odb10. The results indicated that the percentage of complete BUSCOs (C) is 97.9%, reflecting a high level of completeness. The draft chromosome-level genome was 312.42 Mb, including 206 scaffolds, with an N50 of 34.04 Mb and the largest contig size of 41.54 Mb, along with a GC content of 31.69% (Table 1). The genome size of S. kanssuensis is slightly smaller than that of Agrilus biguttatus (368.10 Mb)21, but larger than that of Agrilus cyanescens (292.3 Mb)22. Following Hi-C scaffolding, 98.68% of the genome was anchored to 11 pseudochromosomes, determined based on the chromatin interaction heatmap (Fig. 3). The total length of pseudochromosomes was 309.02 Mb, with individual lengths ranging from 11.70 Mb to 41.54 Mb (Table 2). Among them, the X chromosome was 11.70 Mb in length, comprising 46 contigs.

The chromatin interaction heatmap among 11 chromosomes (A) and circle genome landscape (B) of Sambus kanssuensi. From outside to inside, the circles represent chromosome (a), gene density (b), tandem repeat density (c), GC content (d).
Genome annotation
The repetitive elements of the S. kanssuensis genome were identified using a combination of de novo annotation and homology-based annotation methods. Tandem repeats and interspersed repeats were the predominant repeat sequences in the genomes. Tandem repeat prediction was performed using the software TRF v4.0938 and MISA v2.139. For interspersed repeats, RepeatMasker v4.1.5 (-noLowSimple -pvalue 0.0001; http://repeatmasker.org) was used to align against public databases for homology-based annotation, while LTR_retriever v2.9.840 (-threads 16 -noanno) and RepeatModeler v2.0.541 (-database mydb -threads 16) were employed for de novo annotation. A total of 10.31 Mb of repeat sequences were identified, accounting for 48.05% of the S. kanssuensis genome, which included 36.26% interspersed nuclear elements, 10.52% long terminal repeats and other sequences (Supplementary Table 2). Among the interspersed repeats, retroelements constituted 8.16%, while DNA transposons accounted for 28.1%. Unclassified repeats comprised 3.96% of the total genome.
Both noncoding RNA genes (ncRNAs) and small nuclear RNA genes (snRNAs) were identified in S. kanssuensis genome (Supplementary Table 3). The ncRNAs includes microRNA genes (miRNAs), ribosomal RNA genes (rRNAs), and transfer RNA genes (tRNAs). In this study, miRNAs, snRNAs, and rRNAs were detected using Rfam database (release 13.0)42 and the program Infernal v1.1.443, while tRNAs were predicted using tRNAscan-SE v2.0.12 (-E -j tRNA.gff -o tRNA.result -f tRNA.struct –thread 16). The ncRNAs were annotated using Infernal v1.1.4 (–cut_ga –rfam –nohmmonly –fmt 2) and RNAmmer v1.244 (-S euk -m tsu, lsu, ssu). The numbers of miRNAs, tRNAs, rRNAs, and snRNAs were 53, 1862, 200 and 34, respectively. The rRNAs included 172 large subunit rRNAs (5S, 5.8S and 28S rRNAs) and 28 small subunit rRNAs (18S rRNAs). The tRNAs had 21 isotypes. The snRNAs included 9 CD-box, 3 HACA-box, 21 splicing and 1 scaRNA.
The PCGs were annotated using integrated strategies that combined transcriptome-based prediction, ab initio prediction and homology-based prediction. For transcriptome-based prediction, the full-length transcript data from Oxford Nanopore Technologies were processed using TransDecoder to predict coding frames. PacBio sequences were processed using subreads, and the circular consensus sequences (CCS) reads are identified through CCS in SMRTLink. Then, IsoSeq v3 (https://github.com/PacificBiosciences/IsoSeq) was employed for full-length identification, error correction, and clustering. The error-corrected and redundant-free full-length sequences were aligned using pbmm2, and the results were further refined and reconstructed into transcripts using IsoSeq. For ab initio prediction, the softwares augustus v3.5.045 (–uniqueGeneId = true –noInFrameStop = true –gff3 = on –strand = both), genscan v1.046 and Glimmerhmm v3.0.447 (-f -g) were utilized for gene annotation. For homology-based prediction, miniprot v0.1348 (–gff -Iut50) was used to identified the PCGs of S. kanssuensis based on the known sequences from Coccinella septempunctata, Harmonia axyridis, Tribolium castaneum, and Ulomoides dermestoides. The results from the above method were integrated using EVidenceModeler v1.1.149 to generate the final gene set. Transcriptome annotation was served as expressed sequence tag (EST) evidence, while homology prediction results provided protein homology evidence, and the combined de novo annotation results were used as input for gene prediction. The results revealed that S. kanssuensis genome contains 12,723 PCGs, with 73,788 exons and 61,065 introns (Table 1). The average length of mRNA and coding sequences (CDS) per gene are 13,101.27 bp and 1,468.96 bp, respectively.
Gene functional annotation was carried out by querying the UniProtKB (SwissProt + TrEMBL) databases using Diamond v2.1.850 (–evalue 1e-05). Protein domain and Gene Ontology (GO) annotations were retrieved through eggNOG-mapper v2.0.14551 with the eggNOG v5.0 database, as well as by running InterProScan v5.60-92.04652 against the Pfam53, Smart54, Gene3D v21.055, Superfamily, and Conserved Domains Database (CDD) collections. A total of 11,977 genes were annotated, representing 94.14% of the total genome. The genome contains 9,333 KEGG pathway terms and 9,707 GO items (Table 3). To date, the S. kanssuensis genome is the first genome with both gene annotation and functional annotation in the family Buprestidae.
Data Records
The raw sequencing and genome assembly data of S. kanssuensis were deposited in NCBI. The PacBio, Illumina, Hi-C, and transcriptome data can be found under accession number SRP55981656. The BioProject accession number is PRJNA121300857. The assembly genome is available in NCBI under accession number GCA_047651835.158. Additionally, the data of genome annotation have been deposited in the Figshare database59.
Technical Validation
Assessment of the genome assembly and annotation
The completeness of the chromosome-level genome assembly and annotation was assessed using BUSCO (Supplementary Table 4). The results indicated that the complete BUSCOs (C) were 97.90% for the assembly and 96.10% for the annotation. The duplication rate of genome annotation was 48.05%. The assembled genome was evaluated based on the map rate and coverage, which were calculated using BWA v0.7.1760. The mapping rate and coverage were 99.71% and 96.12%, respectively.
Code availability
No specific programs or codes were used in this study. The specific parameters for the softwares used in this study are described in detail in the methods section.
References
Berenbaum, M. R. The chemistry of defense: theory and practice. Proc. Natl Acad. Sci. USA 92, 2–8 (1995).
Chen, M. S. Inducible direct plant defense against insect herbivores: a review. Insect Sci. 15, 101–114 (2008).
Mithöfer, A. & Boland, W. Plant defense against herbivores: chemical aspects. Annu. Rev. Plant Biol. 63, 431–450 (2012).
Erb, M. & Reymond, P. Molecular interactions between plants and insect herbivores. Annu. Rev. Plant Biol. 70, 527–557 (2019).
Higuchi, Y. & Kawakita, A. Leaf shape deters plant processing by an herbivorous weevil. Nat. Plants 5, 959–964 (2019).
Li, J. B., Jiang, L. Y., Qiao, G. X., Chen, J. An integrative strategy used by the aphid Uroleucon formosanum to counter host sesquiterpene lactone defense: Insights from combined genomic and transcriptomic analysis. Insect Sci. 1–19 (2024).
Heckel, D. G. Insect detoxification and sequestration strategies. In Ulvskov, P. (ed.), Annual Plant Reviews. John Wiley and Sons: Oxford, pp. 77–114 (2014).
Malka, O. et al. Glucosylation prevents plant defense activation in phloem-feeding insects. Nat. Chem. Biol. 16, 1420–1426 (2020).
Dreisbach, D. et al. Spatial metabolomics reveal divergent cardenolide processing in the monarch (Danaus plexippus) and the common crow butterfly (Euploea core). Mol. Ecol. Resour. 23, 1195–1210 (2023).
Kubáň, V., Majer, K. & Kolibáč, J. Classification of the tribe Coraebini Bedel, 1921 (Coleoptera, Buprestidae, Agrilinae). Acta Mus. Moraviae, Sci. Biol. (Brno) 85, 185–287 (2000).
Bellamy, C. L. A World Catalogue And Bibliography Of The Jewel Beetles (Coleoptera: Buprestoidea). Volume 3, Buprestinae: Pteobothrini Throguh Agrilinae: Rhaeboscelina. Pensoft, Sofia, pp. 1265–1921 (2008).
Kubáň, V. Catalog: Buprestidae: genus Sambus. In Löbl I & Löbl D (Eds.) Catalogue Of Palaearctic Coleoptera. Vol. 3. Revised and Updated Edition. Scarabaeoidea, Scirtoidea, Dascilloidea, Buprestoidea And Byrrhoidea. Leiden & Boston: Brill, pp. 549–550 (2016).
Huang, X. Y., Wei, Z. H., Lu, J. W. & Shi, A. M. Mitogenomic analysis and phylogenetic relationships of Agrilinae: Insights into the evolutionary patterns of a diverse buprestid subfamily. PLoS ONE 18, e0291820 (2023).
Ouyang, B. W., Huang, X. Y., Gan, Y. J., Wei, Z. H. & Shi, A. M. Three mitochondrial genomes of Chrysochroinae (Coleoptera, Buprestidae) and phylogenetic analyses. Genes 15, 1336 (2024).
Yoshida, T., Nobuhara, J., Uchida, M. & Okuda, T. Buddledin A, B and C, piscicidal sesquiterpenes from Buddleja davidii Franch. Tetrahedron Lett. 17, 3717–3720 (1976).
Houghton, P. J. et al. Buddlejone, a diterpene from Buddleja albiflora. Phytochemistry 42, 485–488 (1996).
Ren, Y. S. et al. Determination of total triterpenoids in fruit of Buddleja lindleyana Fort. by ultraviolet spectrophotometry. J. Anhui Tradit. Chin. Med. Coll. 31, 78–80 (2012).
Ren, Y. S. et al. Two new 3, 4-secooleanane triterpenoids from Buddleja lindleyana Fort. fruits. Phytochem. Lett. 18, 172–175 (2016).
Wang, B. Q. et al. Research progress on the chemical composition and pharmacological activity of Buddleja plants. Chin. Tradit. Pat. Med. 41, 1664–1653 (2019).
Li, Y. et al. Effects of Buddleja lindleyana Fortune on anaesthesia and serum biochemical indices in crucian carp Carassius auratus. J. Dalian Ocean Univ. 35, 491–495 (2020).
Garland, S. et al. The genome sequence of a jewel beetle, Agrilus biguttatus (Fabricius, 1776). Wellcome Open Res. 9, 413 (2024).
Telfer, M. G. et al. The genome sequence of a metallic wood-boring beetle, Agrilus cyanescens (Ratzeburg, 1837). Wellcome Open Res. 9, 46 (2024).
Coyne, K. J. et al. Improved quantitative real-time PCR assays for enumeration of harmful algal species in field samples using an exogenous DNA reference standard. Limnol. Oceanogr. Methods 3, 381–391 (2005).
Yang, X. et al. HTQC: A fast quality control toolkit for Illumina sequencing data. BMC Bioinformatics 14, 33 (2013).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 7, 764–770 (2011).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Guan, D. F. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Zhang, X. T., Zhang, S. C., Zhao, Q., Ming, R. & Tang, H. B. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585 (2017).
Ou, S. J. & Jiang, N. LTR_retriever: A Highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl Acad. Sci. USA 117, 9451–9457 (2020).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 49, D192–D200 (2021).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Delcher, A. L., Bratke, K. A., Powers, E. C. & Salzberg, S. L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679 (2007).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics 39, btad014 (2023).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Buchfink, B., Xie, C. & Huso, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. methods 12, 59–60 (2015).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Finn, R. D. et al. InterPro in 2017–beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 (2016).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Letunic, I., Khedkar, S. & Bork, P. SMART: recent updates, new developments and status in 2020. Nucleic Acids Res. 49, D458–D460 (2021).
Lewis, T. E. et al. Gene3D: Extensive prediction of globular domains in proteins. Nucleic Acids Res. 46, D1282 (2018).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP559816 (2025).
Sambus kanssuensis isolate:2024703 Genome sequencing. Genbank. https://identifiers.org/ncbi/bioproject:PRJNA1213008 (2025)
Wei, Z. Chromosome-level genome assembly of Sambus kanssuensis (Coleoptera: Buprestidae). GenBank https://identifiers.org/ncbi/insdc.gca:GCA_047651835.1 (2025).
Wei, Z. et al. Chromosome-level genome assembly of Sambus kanssuensis (Coleoptera: Buprestidae). Figshare https://doi.org/10.6084/m9.figshare.28435442 (2025).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Acknowledgements
We extend our gratitude to Ms Bowen Ouyang and Ms Haizhi Wang (China West Normal University) for their assistance in collecting samples. This study was supported by the Sichuan Provincial Natural Science Foundation (2024NSFSC0076), the Natural Science Special (Special Post) Scientific Research Fund Project of Guizhou University (2023-06), and the Qiankehe Platform Talent (BQW[2024]012).
Author information
Authors and Affiliations
Contributions
D.Y. designed the research; A.S. identified the specimens; Z.W., Y.L. and X.C. conducted experiment and data analyses; Y.L. and J.W. collected the specimens; Z.W. and J.L. wrote the original draft; S.D. and D.Y. reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wei, Z., Li, Y., Li, Y. et al. Chromosome-level genome assembly of Sambus kanssuensis (Coleoptera: Buprestidae). Sci Data 12, 895 (2025). https://doi.org/10.1038/s41597-025-05271-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05271-7
