
Abstract
Understanding the spatial distribution of gene expression in the pancreas is essential for establishing the molecular basis of pancreatic function in healthy and disease contexts. Recent platforms offer a robust method for quantifying gene expression within a spatial context. Here, we report spatial transcriptomic profiling from pancreas samples obtained from three donors with type 2 diabetes (T2D) and three donors with normal glucose tolerance (NGT). Our analysis identified a major technical challenge: substantial transcript bleed of highly abundant genes (e.g., INS and GCG) into adjacent tissue regions. We demonstrate that this bleed can be computationally corrected using probabilistic models. Our analysis highlights the importance of incorporating bleed-correction techniques in the preprocessing of spatial transcriptomic profiling data. In summary, this study provides a dataset, methods, and resources to investigate the spatial regulation of gene expression in normal and T2D-affected human pancreas.
Similar content being viewed by others


Background & Summary
The pancreas is a dual-purpose organ with exocrine (digestion) and endocrine functions (hormone secretion for blood glucose regulation)1. Endocrine cell types and functions are localized to pancreatic islets, mini-organs that control glucose homeostasis through tightly regulated secretion of endocrine hormones including insulin and glucagon2. Dysregulated hormone secretion in pancreatic islets is a hallmark of T2D. Spatial mapping of gene expression within the pancreas could improve our understanding of the molecular basis of islet dysfunction leading to T2D.
Historically, studies of spatial trends in gene expression relied on techniques like fluorescence in situ hybridization3. However, such methods have inherent limitations, such as low throughput capacity and a limited number of markers. Recent advances in spatial transcriptomics have greatly increased the resolution and scale for studying spatial gene expression (reviewed in Rao et al.4). These spatial transcriptomics technologies—pairing traditional histology with spatial RNA sequencing5—have enabled exploration into the spatially resolved transcriptional patterns simultaneously across many genes and tissues6,7. To date, few studies using spatial platforms have considered the human pancreas in the normal and diabetic context8. To investigate how spatial patterns change between donors with normal glucose tolerance (NGT) and type 2 diabetes (T2D) (Table 1), we generated a dataset of three NGT and three T2D donors, with replication, using the 10x Genomics Visium Spatial Gene Expression v1 platform.
Methods
Source of human pancreas
Human pancreas samples were obtained from Prodo Laboratories (Aliso Viejo, CA). These samples were isolated from cadaverous donors whose organs were consented for research. As per the National Institutes of Health (NIH) Office of Human Subjects Research Protection (OHSRP) policy, tissue obtained from deceased individuals do not fall under the guidelines of human subject research. All experimental protocols performed for this study were approved under NIH guidelines. Approximately 1.5 cm3 of pancreas tissue was excised from the tail of each cadaveric pancreas. The tissue was dipped in 1% chlorhexidine and washed with saline to remove contaminants. The tissue was then rinsed in Krebs-Ringer solution containing RNase inhibitor (Sigma R7397) and prepared for cryosectioning by embedding them in optimal cutting temperature (OCT) compound, using isopentane cooled at liquid nitrogen temperature. The frozen tissue was placed into a cryovial cooled on dry ice and stored at −80 °C for up to 72 hours before shipping to our laboratory in dry ice. Upon receipt, we placed the frozen sample in a liquid nitrogen tank for long term storage.
Sample preparation, fixation, and staining
We embedded frozen pancreatic tissue samples into OCT compound, solidified in an isopentane ice bath to form individual OCT blocks, and maintained at a temperature of −15 °C to −20 °C for processing or at −80 °C for storage. We sectioned tissue OCT blocks into 10 µm sections and placed them onto Visium Spatial Tissue Optimization Slides (PN-3000394) or Visium Spatial Gene Expression Slides (PN-2000233) as recommended by the manufacturer (10x Genomics, Visium Spatial Protocols - Tissue Preparation Guide CG000240). We fixed and stained Visium slides with hematoxylin and eosin (H&E) according to manufacturer’s instructions (10x Genomics, Methanol Fixation, H&E Staining & Imaging for Visium Spatial Protocols CG000160).
Microscopy for spatial transcriptomics analysis
Following fixation and H&E staining, we imaged the slides using a Leica DMi8 microscope equipped with a CMOS DMC6200 color camera with pixel shift technology (Leica), driven by the Leica LAS X software. We performed image acquisition by employing a HC Plan Apochromatic CS2 10x/0.4 dry lens (Leica) with exposure time of the camera set to 10 ms (consistently for all images) and digital resolution set to 9.2 megapixels (3,840 × 2,400 px, 8 bit). The navigator module of the LAS X software used a tile scanning approach to acquire the entire area of each section, including the fiduciary markers. We captured images as.lef files and exported them as.tif files with lossless compression.
Optimization of tissue permeabilization conditions
After fixation, staining, and imaging, we performed a permeabilization time course followed by fluorescent cDNA synthesis and imaging. We determined an optimal incubation time of 18 minutes for tissue permeabilization (10x Genomics, Visium Spatial Gene Expression Reagent Kits - Tissue Optimization User Guide CG000238) (Fig. 1).

Tissue permeabilization optimization results. Brightfield H&E (top) and fluorescent cDNA (bottom) images from permeabilization time course and fluorescent cDNA synthesis. Image 8 indicates the optimal incubation time of 18 minutes.
Spatial RNA capture, library construction, and sequencing
After staining and imaging, we processed Visium slides to generate spatially barcoded cDNA libraries as per the manufacturer’s instructions (10x Genomics, Visium Spatial Gene Expression Reagent Kits User Guide CG000239; Fig. 2). Briefly, we permeabilized the tissue, releasing mRNA for capture by primers on the Visium slide capture areas. Each capture area was 6.5 × 6.5 mm and contained 4,992 spots of spatial barcodes, where each spot is 55 μm in diameter. We reverse transcribed captured mRNAs into cDNA and coupled them to spatial barcodes during second strand synthesis. We transferred spatially barcoded cDNA sequences from the Visium slide to a tube for library construction. We pooled and sequenced dual-indexed libraries on the NovaSeq platform (Illumina, San Diego, CA, USA) with the following read lengths: Read1 + Index1 + Index2 + Read2 (28 + 10 + 10 + 90), where Index1 and Index2 are the sample indices, Read1 contains the 16 bp Spatial Barcode and 12 bp UMI, and Read2 contains the cDNA insert.

Visium assay workflow. Generation of spatial transcriptomic profiles of human pancreas from normal glucose tolerant (NGT) and type 2 diabetes (T2D) donors. We captured tissue sections with ≥2 replicates onto Visium Spatial Gene Expression slides and performed spatially-resolved RNA sequencing to analyze gene expression patterns within intact tissue sections.
Data Records
We deposited images from the Visium experiments into the Gene Expression Omnibus (GEO) under accession ID GSE2643319. We deposited sequencing data (FASTQs from Illumina NovaSeq) and 10x Genomics Visium Space Ranger output (feature, barcode, and raw count matrices as h5 files) as part of the RNA-seq molecular dataset in the database of Genotypes and Phenotypes (dbGaP) under accession ID phs001188.v3.p110. We deposited cloupe files compatible with 10x Genomics’ Loupe Browser software to Zenodo11.
Sample naming conventions vary across data repositories. To address this, we have provided a key (Table 2) mapping sample IDs to donor/replicate numbering used in this data descriptor.
Technical Validation
Spatial transcriptomic data processing, islet annotation, and quality control procedures
We used Space Ranger (version 2.1.1) with default parameters to process the raw Visium spatial gene expression data and generate gene expression values associated with each spatial location12. The gene expression data consist of gene-specific unique molecular identifier (UMI) counts, measured across thousands of 55 μm diameter spots with spot-specific barcodes on 6.5 × 6.5 mm Visium capture areas. We normalized spot UMI counts using scanpy v1.10.1. Sequencing depth varied from spot-to-spot in ST assays. To address this, we normalized spots by total counts per spot and scaled (counts per 10,000 [CP10k]) using scanpy’s normalize_total function.
To verify tissue integrity and accuracy of spatial capture, we conducted a histological review of H&E stained images to identify major histological features, including islets, vasculature, and ducts (Fig. 3a). We compared the identified islet regions to the expression patterns of islet marker genes (e.g., INS for beta cells that reside in islets). We found a high overlap of the islet region labelled from H&E image review and the regions with the highest INS expression, validating the accurate identification of islets within the pancreatic tissue samples (Fig. 3a,b).

Representative sample expression and quality controls. (a) Pathologist annotations overlaid on H&E histology slide, with islet regions circled in red, ducts/ductules in orange, and vascular structures in yellow. (b) INS expression. (c) Number of Visium barcoded spots covered by the tissue sample from each donor.
For each sample, our dataset contained two to four tissue slices. We evaluated the number of spots per tissue as an indirect measure of the area of each section (Fig. 3c). We found consistent results across replicates, in line with our expectations as these samples were sequential or near-sequential sections (Fig. 4).

Dataset quality metrics returned by 10X Genomics Space Ranger Pipeline. Quality metrics from 10x Genomics Visium Space Ranger for each processed tissue section from 6 donors, 2–4 replicates each. Replicates are sequential sections or near-sequential sections.
Identification and cleaning of transcript bleed artifacts
While the spatial distribution of INS expression was largely concordant with manually annotated islets from H&E images (Fig. 3a,b), visual inspection of INS expression revealed substantial levels of insulin transcripts across the entire tissue section, including exocrine regions. This observation suggested a degree of transcript diffusion during sample preparation (Fig. 3b). To correct for such transcript bleed, we used a computational approach that uses probabilistic models to adjust spatial gene expression data for transcript bleed (SpotClean13 (version 1.4.1)) (Fig. 5).

Transcript bleed correction. (a) INS expression overlaid on H&E histology slide. (b) Bleed-corrected INS expression.
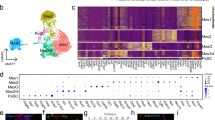
After transcript bleed-correction, cell type marker genes for endocrine and exocrine cell types were more cleanly divided into distinct compartments, which more faithfully reflected established understanding of human islets as distinct endocrine cell clusters, delineated by a basement membrane, surrounded by primarily exocrine tissue (dominated by acinar cells) (Fig. 6)14. These findings indicated that bleed correction can validate the quality of spatial transcriptomic datasets, and users who access the deposited raw data can replicate this result by following our methods.

Cell type marker gene expression. Visualization of bleed-corrected gene expression of cell type marker genes. Marker genes include CD68 (macrophage), COL1A1 (endothelial), GCG (alpha), INS (beta), KRT19 (ductal), PPY (gamma), PRSS1 (acinar), and SST (delta)14.
Concluding remarks
In summary, this dataset offers spatial transcriptomic profiles of the human pancreas from both NGT and T2D donors, at 55 μm resolution. Our initial observations of these profiles identified substantial bleed of abundant islet genes, but probabilistic computational correction mitigated this technical artifact effectively.
Limitations of these data include the limited spatial resolution of the Visium Spatial Gene Expression v1 assay and the limited number of donors profiled. The Visium v1 platform spatially barcodes transcripts in a 6.5 × 6.5 mm capture area covered by 4,992 spots, each 55 μm in diameter. At this resolution, data from each spot represented transcriptomes from multiple cells. Recently released assays improve resolution to a near single-cell level12.
While exploratory analyses of transcriptional changes in T2D pancreas are a primary application of these data, a larger sample size will be required to overcome donor-to-donor variation when making these comparisons. Having demonstrated the integrity of these data, they may be combined and meta-analyzed with future data sets towards this aim. Moreover, these data alone may be suitable for targeted analyses, including validating the presence or spatial distribution of genes of interest. Notably, since these spatial profiles represent direct transfer of RNA from fresh frozen pancreas and are not dependent on pre-defined human transcriptome probe sets, this resource may be used to detect unannotated and/or non-coding transcripts of interest, which may not be detectable with probe-based RNA assays or protein-based assays (e.g., immunofluorescence imaging) of fixed tissue. Third-party software and frameworks enable continued exploration of spatial patterns across tissues in Visium v1 data. Deep learning models15 can improve resolution by predicting transcript counts in inter-spot space on a capture slide. Computational methods, like those provided by the Spatial-eXpression-R (spacexr) library16,17, can deconvolute cell types in spots and predict differential expression across spatial axes. Paired with tools like these, the spatially resolved transcriptional profiles of human pancreas presented here may yield additional insights about healthy and T2D pancreas biology.
Code availability
The code used for data processing and bleed correction are available at https://github.com/CollinsLabBioComp/publication-visium_preprocessing.
References
Leung, P. S. Overview of the pancreas. Adv. Exp. Med. Biol. 690, 3–12 (2010).
Campbell, J. E. & Newgard, C. B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 22, 142–158 (2021).
Simonis, M. & de Laat, W. FISH-eyed and genome-wide views on the spatial organisation of gene expression. Biochim. Biophys. Acta 1783, 2052–2060 (2008).
Rao, A., Barkley, D., França, G. S. & Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 596, 211–220 (2021).
Spatial Gene Expression - 10x Genomics. https://www.10xgenomics.com/products/spatial-gene-expression.
Rao, N., Clark, S. & Habern, O. Bridging genomics and tissue pathology. Genetic Engineering & Biotechnology News 40, 50–51 (2020).
Ståhl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016).
Cui Zhou, D. et al. Spatially restricted drivers and transitional cell populations cooperate with the microenvironment in untreated and chemo-resistant pancreatic cancer. Nat. Genet. 54, 1390–1405 (2022).
Bonnycastle, L. L. et al. GEO. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE264331 (2024).
Boehnke, B. et al. The Finland-United States Investigation of NIDDM Genetics (FUSION) Study - Islet Expression and Regulation by RNAseq and ATACseq. dbGaP https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001188.v3.p1.
Howell, N. et al. A spatial transcriptomics dataset of pancreas sections in normal glucose tolerance and type 2 diabetic donors. Zenodo https://doi.org/10.5281/zenodo.15177311 (2025).
Spatial Transcriptomics - 10x Genomics. https://www.10xgenomics.com/spatial-transcriptomics.
Ni, Z. et al. SpotClean adjusts for spot swapping in spatial transcriptomics data. Nat. Commun. 13, 2971 (2022).
van Gurp, L. et al. Generation of human islet cell type-specific identity genesets. Nat. Commun. 13, 2020 (2022).
Monjo, T., Koido, M., Nagasawa, S., Suzuki, Y. & Kamatani, Y. Efficient prediction of a spatial transcriptomics profile better characterizes breast cancer tissue sections without costly experimentation. Sci. Rep. 12, 4133 (2022).
Cable, D. M. et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 40, 517–526 (2022).
Cable, D. M. et al. Cell type-specific inference of differential expression in spatial transcriptomics. Nat. Methods 19, 1076–1087 (2022).
Acknowledgements
NIH NIAMS Genomic Technology Section supported sequencing efforts for this study. This research was supported by the US National Institutes of Health grants ZIAHG000024 (to F.S.C. and L.G.B.), K99DK13917501 (to D.L.T.).
Funding
Open access funding provided by the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
L.L.B., C.C.R., D.L.T. and F.S.C. designed the research. L.L.B., A.J.S., C.M.G., M.R.E. and D.R. generated data for this study. N.H., Z.W., N.N., N.S., C.D., C.C.R. and D.L.T. analyzed data. L.L.B., C.C.R., D.L.T., L.G.B. and F.S.C. supervised the study. N.H., Z.W., C.C.R. and D.L.T. wrote the paper. All authors reviewed and approved the paper.
Corresponding authors
Ethics declarations
Competing interests
L.G.B. is a member of the Illumina Medical Ethics Board, receives research support from Merck, Inc, and royalties from Wolters-Kluwer.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Howell, N., Weiss, Z., Bonnycastle, L.L. et al. A spatial transcriptomics dataset of pancreas sections in normal glucose tolerance and type 2 diabetic donors. Sci Data 12, 1526 (2025). https://doi.org/10.1038/s41597-025-05450-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05450-6
