
Abstract
Spinibarbus caldwelli is an economically important freshwater species within the Cyprinidae family, abundant in the middle and lower reaches of the Yangtze River and its adjacent basins. As a promising species suitable for aquaculture in southern China, the lack of genomic resources has hampered the genetic breeding and conservation. Here, we release a chromosome-level genome assembly for S. caldwelli using PacBio HiFi long-reads, Illumina short-reads, and Hi-C sequencing data. The final genome assembly is 1.77 Gb in size, with a contig N50 of 24.27 Mb. Using Hi-C scaffolding, 99.14% of the contigs were successfully anchored to 50 chromosomes, resulting in a scaffold N50 of 35.29 Mb. The final genome assembly shows a BUSCO completeness of 98.27%. The assembled genome contains 49.41% repetitive sequences and 51,505 predicted genes, 90.83% of which have been functionally annotated. This genome provides a genetic basis for S. caldwelli, facilitating the exploration of Cyprinid phylogeny, genetic improvement, and conservation efforts.
Similar content being viewed by others


Background & Summary
Spinibarbus caldwelli, which belongs to the family Cyprinidae, subfamily Barbinae, is native to the middle and lower reaches of the Yangtze River and its associated river system in China, Laos and Vietnam. This species specifically inhabits clear-water, gravel-bottomed river segments1,2, making it an indicator species for assessing environmental health in freshwater ecosystems. As an economic freshwater fish, S. caldwelli is regarded as ideal for pond and large-surface water aquaculture. With the spread of domestication and breeding techniques, S. caldwelli has become a key species in aquaculture in eastern and southern China3. However, since the 1980s, overfishing, environmental pollution, and water conservancy projects have led to a sharp decline in the wild population size of S. caldwelli3.
Yuan et al. investigated the genetic diversity and population structure of S. caldwelli using mitochondrial DNA gene as molecular markers4. Tang et al. found significant genetic differentiation among the wild populations, mainly due to geographical isolation or human activities5. Ai et al. sequenced and assembled the complete mitochondrial genome of S. caldwelli, which exhibits a gene composition, arrangement, and transcriptional direction identical to those of most vertebrates6. Tang et al. studied the vasa homologs in S. caldwelli and provided insights into the molecular mechanism of germ cell development and differentiation7. Previous studies were mainly based on mitochondrial genes or particular gene cloning in S. caldwelli. However, a high-quality chromosome-level genome assembly remains unavailable, limiting the sustainable development and utilization of S. caldwelli.
Here, we employed a combined strategy using Illumina, PacBio, and Hi-C technologies to generate sequencing data for S. caldwelli genome assembling. The assembled genome size was 1.77 Gb with contig and scaffold N50 reaching 24.27 Mb and 35.29 Mb, respectively, demonstrating excellent genome integrity and sequence continuity. The annotated genome contains 49.41% repetitive sequences, 51,505 predicted genes, and 90.83% of these genes were functionally annotated. Synteny analysis showed that S. caldwelli and S. sinensis shares highly genome collinearity, indicating minor difference in chromosome karyotype. The construction of genomic resources for S. caldwelli provides support for elucidating the genetic basis of important traits and facilitates ecological conservation through the establishment of germplasm resource banks, promoting the sustainable development of S. caldwelli.
Methods
Sample collection
A female S. caldwelli (Fig. 1A) was collected from the Taojiang River (25.631087°N, 115.014197°E) in Ganzhou City, Jiangxi Province, China. Genomic DNA was extracted from muscle tissue and used for Illumina short-read, PacBio long-read, and Hi-C sequencing. RNA was extracted from various tissues, including scale, skin, fin, muscle, gill, liver, intestine, gonad, heart, bladder, head kidney, eye, brain, intermuscular bone, spleen, and embryo. The extracted RNA was then pooled into four samples for RNA sequencing. All tissue samples were immediately frozen in liquid nitrogen and stored at −80 °C.

Illustration and karyotype of S. caldwelli. (A) An illustration of S. caldwelli. Scale bar equals 1 cm. (B) Karyotype of S. caldwelli. Scale bar equals 10 μm.
Karyological analysis
Chromosome preparations from the head kidney were made using established methods8,9. Live fish were treated with colchicine, then incubated in hypotonic KCl before fixation in ethanol-acetic acid. Chromosomes were stained with Giemsa and analyzed with KaryoType software10. Chromosomes were categorized based on Levan et al.11. The karyotype of S. caldwelli shows a chromosome complement of 2n = 100, confirming consistency with reference genome assembly data (Fig. 1B). The karyotype of S. caldwelli contains 9 pairs of metacentric (m), 16 pairs of submetacentric (sm), 15 pairs of sub-telocentric (st), and 10 pairs of telocentric (t) chromosomes. No specific sex chromosomes were observed (Fig. 1B).
Library preparation and genome survey
For genome survey purposes, 1–1.5 μg of genomic DNA from S. caldwelli was randomly fragmented using a Covaris system. The resulting fragments were purified and size-selected to an average of 200–400 bp using the Agencourt AMPure XP-Medium kit (Beckman Coulter, Inc., CA, USA). DNA fragments were processed through end-repair, 3’adenylation, adapter ligation, and PCR amplification, followed by purification using the AxyPrep Mag PCR Clean-up Kit (Axygen, Hangzhou, China). The double-stranded PCR products were heat-denatured and circularized using a splint oligo sequence to generate single-stranded circular DNA, which was formatted as the final library and verified by quality control. The prepared library was sequenced on the Illumina NovaSeq 6000 platform. After acquiring 56.96 Gb of raw sequence data, paired-end raw reads were quality-filtered using fastp (v 0.20.0)12 to remove low-quality reads, adapters, and poly-N sequences. Contamination was checked by aligning 100,000 random reads to the NT database.
PacBio and Hi-C based whole-genome sequencing
SMRTbell target size libraries were constructed for sequencing according to standard protocol of PacBio (Pacific Biosciences, CA, USA) using 15 kb preparation solutions. Sheared gDNA underwent DNA damage repair, blunt-end ligation with hairpin adapters, exonuclease treatment, and size selection. Libraries were purified and validated using Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). Sequencing was performed on the PacBio Sequel II platform and raw polymerase reads were processed via SMRT Link v8.0 for adapter trimming and quality filtering.
Hi-C libraries were constructed from genomic DNA of S. caldwelli. Optimized formaldehyde crosslinking preserved chromatin conformation, followed by biphasic lysis and DpnII digestion with NEB buffer and BSA. Biotin-14-dATP facilitated fill-in labeling for capture. DNA was purified, sheared to ~400 bp, and prepared using the NEBNext Ultra II Kit13 (New England Biolabs, MA, USA). Hi-C sequencing on the Illumina NovaSeq 6000 platform produced 176.53 Gb of raw data, comprising 370 million paired-end reads.
Finally, we obtained high-quality sequencing data comprising 63.07 Gb of PacBio HiFi reads (~35.20×), 56.96 Gb of Illumina reads (32.01×), and 176.53 Gb of Hi-C data (42.03×), providing robust genomic coverage for downstream analysis (Table 1).
Genome size estimation and de novo genome assembling
To characterize the genomic features of S. caldwelli, k-mer analysis was performed on the Illumina sequencing data prior to assembly. Quality-filtered reads underwent 17-mer frequency analysis via the KMC14 tool. Genome size was estimated using G = K-num/K-depth, analyzing 17-mer depth distribution from cleaned 350-bp library reads via gce15 and FindGSE16. The estimated genome size is 1,609,818,105.00 bp with a heterozygosity of 0.80%.
After obtaining the PacBio subreads, the genome was de novo assembled into contigs using the overlap-layout-consensus (OLC) algorithm implemented in Falcon17. All PacBio SMRT reads were then aligned to the assembled contigs using BLASR18, and Quiver19 was employed to correct sequencing errors based on the alignments, using default parameters. To further improve base-level accuracy, the contigs were polished with Nextpolish2 (v 0.2.1)20, incorporating Illumina short reads under default settings. Finally, to remove potentially redundant contigs and generate a non-redundant primary assembly, similarity-based filtering was performed with thresholds of identity ≥0.8 and overlap ≥0.8. This process resulted in a preliminary monoploid assembly of 1.77 Gb, with a contig N50 length of 24.27 Mb, representing a high-quality draft genome.
To achieve chromosome-level assembly, Hi-C reads were first aligned to the draft genome of S. caldwelli using Bowtie2 (v 2.3.2)21. Valid interaction pairs were identified and retained by HiC-Pro (v 2.8.1)22 from uniquely mapped paired-end reads, while invalid pairs, such as dangling ends, self-cycles, re-ligations, and dumped products, were filtered out. The processed reads were then analyzed with LACHESIS23 to cluster, order, and orient the contigs, thereby facilitating the construction of a chromosome-level assembly. The diploid chromosome number of S. caldwelli (2n = 100) informed the scaffolding assembly process (Fig. 2A). Detailed chromosomal statistics and their alignment to the assembled genome are provided in Table 2. After Hi-C-assisted scaffolding, the final assembly yielded 50 chromosome-scale scaffolds, corresponding to the 50 haploid chromosomes of S. caldwelli, and a total of 136 scaffolds overall. The final genome size of S. caldwelli was 1.76 Gb, with a scaffold N50 of 35.29 Mb and a contig N50 of 23.94 Mb (Table 3).

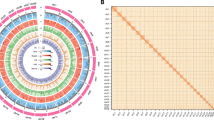
Features of S. caldwelli genome assembly. (A) The genomic landscape of S. caldwelli. The circus plot from the outer to inner refers to chromosome (chr01A-chr25B) (a), gene density (b), guanine-cytosine (GC) content (c), tandem repeats abundance (d), repeat elements abundance (e), and ncRNA abundance (f), respectively. (B) Hi-C interaction heatmap of S. caldwelli genome assembly.
Genome annotation
A de novo specific repeat library for S. caldwelli was constructed using RepeatModeler (v 1.0.11)24 LTR sequences were predicted and deduplicated using LTR_FINDER_parallel25 and LTR_retriever (v 2.9.0)26. The two libraries were merged to create a TE library file (TE.lib). The genome was masked once, replacing repetitive sequences with “N”, and a subsequent de novo search for repetitive sequences was conducted using RepeatModeler to generate a de novo library file (RepMod.lib).
RepeatMasker (v 1.331)27 was used to identify and mask repeat elements in the S. caldwelli genome using a custom library. Analysis revealed that approximately 42.75% of the genome consists of repetitive elements, including LTR elements (6.81%), DNA transposons (23.78%), LINE elements (6.44%), and simple repeats (0.38%). The distribution of these elements, including simple repeats and transposable elements (TEs), was mapped across each chromosome (Fig. 3A).

The repeat elements distribution and identified protein-coding genes in the S. caldwelli genome. (A) Distribution of divergence rate for transposable elements in the S. caldwelli genome. (B) Venn diagram showing the number of shared and unique genes annotated with different databases in the S. caldwelli genome.
Non-coding RNAs (ncRNAs) and transfer RNAs (tRNAs) in S. caldwelli were identified using Infernal (v 1.1.2)28 for ncRNAs and tRNAscan-SE (v 2.0)29 for tRNAs. The analysis detected a total of 39,459 ncRNAs in the S. caldwelli genome, including 4,186 rRNAs, 5,419 small RNAs, 13,502 regulatory RNAs and 16,352 tRNAs. (Revision Note: The numbers of ncRNAs and tRNAs have been corrected in the revised manuscript, due to an earlier misstatement).
Gene prediction was performed on the repeat-masked genome using three independent approaches: reference-guided transcriptome assembly, ab initio prediction, and homology-based annotation.
For RNA-seq–based prediction, RNA-seq datasets from four libraries were first quality-checked using FastQC30, aligned to the genome using STAR (v 2.7.3a)31, and assembled into transcripts with Stringtie (v 1.3.4 d)32. Open reading frames (ORFs) within the assembled transcripts were predicted using PASA (v 2.3.3)33.
For ab initio prediction, RNA-seq reads were also de novo assembled using StringTie and analyzed with PASA to generate a gene structure training set. This training set was used to guide Augustus (v 3.3.1)34 under default parameters. GeneMark-S35 was used to support unsupervised training of gene models based on transcript evidence.
For homology-based annotation, protein sequences from the NCBI and NGDC database for Carassius auratus (GCF_003368295.1), Danio rerio (GCF_000002035.6), Megalobrama amblycephala (GCF_018812025.1), Sinocyclocheilus anshuiensis (GCF_001515605.1), Sinocyclocheilus grahami (GCF_001515645.1), Sinocyclocheilus rhinocerous (GCF_001515625.1), Ctenopharyngodon idella (GCF_019924925.1) and Spinibarbus sinensis (CRA008955)36. These sequences were then mapped to the S. caldwelli genome using GeMoMa37 (Table 4).
All gene models derived from the three approaches were integrated using EVidenceModeler (EVM) with default parameters. Confidence weights were assigned in the order: PASA > GeMoMa > ab initio. Genes containing transposable elements (TEs) were identified and removed using TransposonPSI38. Additionally, miscoded genes, such as those with frameshift mutations or internal stop codons, were filtered out. Untranslated regions (UTRs) and alternative splicing isoforms were annotated using PASA, and only the longest isoform per locus was retained as the representative transcript.
Functional annotation of protein-coding genes was performed by aligning the predicted sequences against NR39, SwissProt40, KEGG41, KOG42 databases using Blastp (v 2.7.1)43 and InterProScan (v 5.32–71.0)44 for GO45 databases. Gene motifs and functional domains were identified via InterProScan, and GO terms were assigned based on matches to InterPro or UniProt entries. The average gene and CDS length were 18.33 kb and 1.60 kb, respectively, and the average number of exons per gene was 9.76. A total of 51,505 protein-coding genes were predicted, of which 46,782 (90.83%) were successfully functionally annotated through these integrated databases and tools (Table 5 & Fig. 3B).
Data Records
The raw sequencing data reported in this study have been deposited in the NCBI Sequence Read Archive (SRA) database under the accession numbers, SRR3120416246, SRR3120416347, SRR3107819448, SRR3107819549, SRR3107819650, SRR3107819851 and SRR3107819952, SRR3301485353. The assembled genome of S. caldwelli is available in GenBank under the accession number GCA044721935.154. Two whole-genome sequencing datasets employing long-read and short read (SRR31204162 and SRR31078198) were generated using the PacBio Sequel II and Illumina HiSeq X Ten platforms, respectively. Two Hi-C datasets (SRR31204163 and SRR31078199) were generated on the Illumina NovaSeq 6000 platform. Four RNA-seq datasets (SRR31078194, SRR31078195, SRR31078196, and SRR33014853) were generated on the Illumina HiSeq X Ten platform. The genome annotation results have been deposited in the figshare database (https://doi.org/10.6084/m9.figshare.26494309)55.
Technical Validation Quality
Quality evaluation of the genome assembly and annotation
The completeness of the genome assembly of S. caldwelli was evaluated using BUSCO56 and CEGMA57. BUSCO analysis was performed using the vertebrata_odb10 dataset, revealing that 99.14% (3,325 out of 3,354) of the expected orthologous genes were present as complete, of which 22.15% were single-copy and 76.98% were duplicated. Only 0.27% were fragmented, and 0.60% were missing (Table 6), indicating a highly complete assembly. CEGMA identified 240 of the 248 core eukaryotic genes (96.77%), including 208 complete genes (83.87%), indicating a high level of completeness in conserved genomic elements.
To assess the base-level accuracy of the genome assembly, Illumina paired-end reads were aligned to the assembled genome using BWA58, and alignment statistics were analyzed with SAMtools31 and BCFtools59. The sequencing reads showed a high mapping rate of 99.95%, covering 99.49% of the genome at 1 × coverage.
For chromosome quality evaluation, strong interactive signals were observed along the diagonals of Hi-C heatmaps, with no significant noise detected in other areas (Fig. 2B), supporting the accuracy of the chromosome assembly.
The quality of the gene prediction was also evaluated using BUSCO, which showed that 98.27% (3,296 out of 3,354) of conserved genes were present in the predicted gene set of S. caldwelli, suggesting high gene annotation completeness. These results indicate a comprehensive and functionally informative annotation set for S. caldwelli.
Chromosomal synteny analysis
A genomic synteny analysis was performed between S. caldwelli and S. sinensis to evaluate their structural characteristics and validate the accuracy of our genome assembly. Similar gene pairs and syntenic blocks were identified and visualized using Mummer (v 4)60 and R (v 4.4.1). The analysis revealed a high degree of collinearity between the two assemblies, with most chromosomes in S. caldwelli retaining their structure compared to S. sinensis (Fig. 4). This high level of consistency suggests the high quality of the assembled and annotated genomes.

Genomic synteny analysis between S. caldwelli and S. sinensis. Sc01-Sc50 represent chromosome 1–50 of S. caldwelli. Ss01-Ss50 represent chromosome 1–50 of S. sinensis.
Code availability
No specific code was developed for this study. Data analyses were performed according to the manuals and protocols provided by the developers of the relevant bioinformatics tools, as described in the Methods section, including the versions used.
References
Zou, P. et al. Preliminary study on the age and growth of Spinibarbus caldwelli. Sichuan Journal of Zoology 3, 26, https://doi.org/10.3969/j.issn.1000-7083.2007.03.007 (2007).
Fang, T. et al. Nutritional composition analysis and quality evaluation of Spinibarbus caldwelli cultured in spring flowing water. Fisheries Science & Technology Information 52, 49–55, https://doi.org/10.16446/j.fsti.20231200227 (2025).
Tuo, Y. et al. Analysis of Natural Selection of Immune Genes in Spinibarbus caldwelli by Transcriptome Sequencing. Frontiers in Genetics 11, 714, https://doi.org/10.3389/fgene.2020.00714 (2020).
Tang, Q. et al. Molecular and morphological data suggest that Spinibarbus caldwelli (Nichols) (Teleostei: Cyprinidae) is a valid species. Ichthyol Research 52, 77–82, https://doi.org/10.1007/s10228-004-0259-x (2005).
Yuan, X. et al. Genetic Structure of Spinibarbus caldwelli Based on mtDNA D-Loop. Agricultural Sciences 10, 173–180, https://doi.org/10.4236/as.2019.102015 (2019).
Ai, W., Peng, X., Huang, X., Xiang, D. & Chen, X. Complete mitochondrial genome of Spinibarbus caldwelli (Cypriniformes, Cyprinidae). Mitochondrial DNA 26, 131–132, https://doi.org/10.3109/19401736.2013.815171 (2013).
Tang, L.-hua et al. cDNA cloning and expression analysis of a vasa-like gene in Spinibarbus caldwelli. Journal of Fisheries of China 36, 868–878, https://doi.org/10.3724/SP.J.1231.2012.27791 (2012).
Blanco, D. R. et al. A new technique for obtaining mitotic chromosome spreads from fishes in the field. Journal of fish biology 81, 351–357, https://doi.org/10.1111/j.1095-8649.2012.03325.x (2012).
Ojima, Y. & Kurishita, A. A new method to increase the number of mitotic cells in the kidney tissue for fish chromosome studies. Proceedings of the Japan Academy, Series B 56, 610–615, https://doi.org/10.2183/pjab.56.610 (1980).
Altınordu, F., Peruzzi, L., Yu, Y. & He, X. A tool for the analysis of chromosomes: KaryoType. Taxon 65, 586–592, https://doi.org/10.12705/653.9 (2016).
Levan, A., Fredg, A. & Sandberg, A. Nomenclature for centromeric position on chromosomes. Hereditas 52, 201–220, https://doi.org/10.1111/j.1601-5223.1964.tb01953.x (1964).
Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2, 107, https://doi.org/10.1002/imt2.107 (2023).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Deorowicz, S. et al. KMC 2: fast and resource-frugal k-mer counting. Bioinformatics 31, 15, https://doi.org/10.1093/bioinformatics/btv022 (2015).
Liu, B et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv preprint arXiv: 1308, 2012, https://doi.org/10.48550/arXiv.1308.2012 (2013).
Sun, H. et al. FindGSE: Estimating genome size variation within human and Arabidopsis using k -mer frequencies. Bioinformatics 34, 550–557, https://doi.org/10.1093/bioinformatics/btx637 (2018).
Chin, C. S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nature methods 13(12), 1050–1054, https://doi.org/10.1038/nmeth.4035 (2016).
Chaisson, M. J. & Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13, 238, https://doi.org/10.1186/1471-2105-13-238 (2012).
Chin, C. S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature methods 10(6), 563–569, https://doi.org/10.1038/nmeth.2474 (2013).
Jiang, H. et al. NextPolish2: A Repeat-aware Polishing Tool for Genomes Assembled Using HiFi Long Reads. Genomics, Proteomics & Bioinformatics 22, 1, https://doi.org/10.1093/gpbjnl/qzad009 (2024).
Langmead, B. & Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, https://doi.org/10.1038/nmeth.1923 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16, 259, https://doi.org/10.1186/s13059-015-0831-x (2015).
Burton, J. N. et al. Chromosome-scale contiguity of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 31, 1119–1125, https://doi.org/10.1038/nbt.2727 (2013).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. PNAS 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35, 265–268, https://doi.org/10.1093/nar/gkm286 (2007).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18, https://doi.org/10.1186/1471-2105-9-18 (2008).
Bedell, J. A., Korf, I. & Gish, W. MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics 16, 1040–1041, https://doi.org/10.1093/bioinformatics/16.11.1040 (2000).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964, https://doi.org/10.1093/nar/25.5.955 (1997).
Andrews, S. FastQC: A quality control tool for high throughput sequence data. Bioinformatics 15, 1968–1971, https://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20, 278, https://doi.org/10.1186/s13059-019-1910-1 (2019).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, 7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Stanke, M. et al. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res 43, 78, https://doi.org/10.1093/nar/gkv227 (2015).
Xu, M. R. X. et al. Maternal dominance contributes to subgenome differentiation in allopolyploid fishes. Nat Commun 14, 8357, https://doi.org/10.1038/s41467-023-28117-0 (2023).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic acids research 44(9), e89, https://doi.org/10.1093/nar/gkw092 (2016).
Urasaki, N. et al. Draft genome sequence of bitter gourd (Momordica charantia), a vegetable and medicinal plant in tropical and subtropical regions. Dna Research 24(1), 51–58, https://doi.org/10.1093/dnares/dsw047 (2017).
Koonin, E. V. et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. J. Genome Biol 5, 1–28, https://doi.org/10.1186/gb-2004-5-2-r7 (2004).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Research 27, 49–54, https://doi.org/10.1093/nar/27.1.49 (1999).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30, https://doi.org/10.1093/nar/28.1.27 (2000).
Galperin, M. Y. et al. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res 43, 261–269, https://doi.org/10.1093/nar/gku1223 (2015).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421, https://doi.org/10.1186/1471-2105-10-421 (2009).
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848, https://doi.org/10.1093/bioinformatics/17.9.847 (2001).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nature genetics 25, 25–29, https://doi.org/10.1038/75556 (2000).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR31204162 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR31204163 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR31078194 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR31078195 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR31078196 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR31078198 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR31078199 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR33014853 (2024).
NCBI GenBank. https://identifiers.org/ncbi/insdc.gca:GCA_044721935.1 (2024).
Wen, L. The genome annotation of Spinibarbus caldwelli. Figshare https://doi.org/10.6084/m9.figshare.26494309 (2024).
Simao, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23(9), 1061–1067, https://doi.org/10.1093/bioinformatics/btm071 (2007).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26(5), 589–595, https://doi.org/10.1093/bioinformatics/btp698 (2010).
Danecek, P. & McCarthy, S. A. BCFtools/csq: haplotype-aware variant consequences. Bioinformatics 33(13), 2037–2039, https://doi.org/10.1093/bioinformatics/btx100 (2017).
Marçais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLOS Computational Biology 14, 1005944, https://doi.org/10.1371/journal.pcbi.1005944 (2018).
Acknowledgements
This work was supported by National Natural Science Foundation of China (32260913), Aquaculture Breeding Joint Research Project of Jiangxi Province (JXNY202207), Dynamic Changes in Population Resource and Reproduction Scale of Spinibarbus Species after Jingdezhen Water Control Project (NCUCQYJY-HX-2024-4) and Research Project on the Protection and Utilization of Germplasm Resources of Spinibarbus caldwelli (gsnz2023-30).
Author information
Authors and Affiliations
Contributions
W.Z. and Y.H. conceived, designed and supervised the study. Y.F. and Q.Z. provided the materials for sequencing. W.Z., S.J. and Z.F. prepared the samples for sequencing. W.Z., Z.F., W.L. and J.S. performed the genome assemble and annotation. Z.F., W.L. and L.F. prepared the figures and tables. Z.F. and W.L. drafted the manuscript, W.Z. and Y.H. revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fan, Z., Lei, W., Fang, Y. et al. Chromosome-level genome assembly and annotation of Spinibarbus caldwelli. Sci Data 12, 1138 (2025). https://doi.org/10.1038/s41597-025-05469-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05469-9
