
Abstract
Here we present the first high-quality chromosome-level genome assembly for Gibbotettix parvipulvillus, a Chinese endemic pygmy grasshopper serving as a significant bioindicator for fragile forest ecosystems and exhibiting unique evolutionary adaptations to restricted microhabitats. Combining 47.43 Gb of PacBio HiFi, 47.32 Gb of Illumina, and 68.34 Gb of Hi-C sequencing data, we achieved an assembly size of 1.42 Gb with exceptional contiguity (contig N50: 100.63 Mb; scaffold N50: 308.03 Mb). The assembly anchored 97.88% of sequences to 7 pseudochromosomes, with chromosome 7 identified as the X chromosome through reduced sequencing coverage (18.04 × versus ~ 34 × for autosomes). Repeat annotation revealed repetitive elements comprise 58.79% of the genome, with LINEs being most abundant (14.15%). We annotated 17,220 protein-coding genes with functional information assigned to 93.82% of them. The assembly demonstrates high completeness (BUSCO score: 98.8% complete genes) and accuracy (read mapping rate > 98.5%). This genomic resource will facilitate research on the evolution of winglessness, adaptation to restricted habitats, and conservation genomics of this ecologically valuable bioindicator taxon.
Similar content being viewed by others


Background & Summary
Species within Gibbotettix (Orthoptera, Caelifera, Tetrigidae, Scelimeninae) exhibit distinctive morphological characteristics, including remarkably rough body surfaces, prominently elevated pronotum forming a humpback-like structure in the anterior half, and the complete absence of both tegmina and hind wings1. This wingless condition represents an intriguing evolutionary adaptation that directly impacts dispersal capabilities and likely contributes to their restricted distribution patterns2. Most Gibbotettix species are endemic with highly localized distributions across China, representing classic examples of species with limited geographic ranges3.
The ecological requirements of Gibbotettix are notably stringent, as they are exclusively found in cool, undisturbed forest environments with specific temperature ranges1,3. Their dietary preferences for mosses and humus, combined with their limited mobility and specific habitat requirements, position them as valuable bioindicators of environmental health4. The presence and abundance of Gibbotettix species could potentially contribute to biodiversity assessment, as arthropod communities are known to respond to environmental pressures and habitat changes. The disjunct distribution pattern observed in Gibbotettix species presents a compelling framework for investigating evolutionary processes. These isolated populations likely represent relict distributions shaped by historical climate fluctuations, geological events, and subsequent adaptation to local conditions5,6. This fragmented distribution also raises fundamental questions regarding speciation mechanisms and genetic diversity maintenance7.
Despite their ecological importance and evolutionary significance, comprehensive genomic studies of Tetrigidae, particularly of the genus Gibbotettix, have been notably absent from the scientific literature8,9. Previous research has primarily focused on morphological taxonomy and limited ecological observations, with molecular analyses largely restricted to mitochondrial markers for phylogenetic placement10,11,12. This significant knowledge gap hampers our understanding of evolutionary processes in this group and limits insights into adaptation mechanisms that enable survival in specific microhabitats.
The advent of high-throughput sequencing technologies has revolutionized our ability to investigate non-model organisms13,14. Chromosome-level genome assemblies provide unprecedented opportunities to explore genome architecture, adaptive evolution, and population genomics of specialized taxa15,16. Such high-quality genomic resources are essential for understanding how environmental specialists like Gibbotettix have evolved their distinctive traits and ecological requirements.
Here, we present the first chromosome-level genome assembly of Gibbotettix parvipulvillus, a representative species of this ecologically important genus. Our assembly spans 1.42 Gb with 97.88% of sequences anchored to 7 pseudochromosomes, including the identification of the X chromosome through coverage analysis. The assembly demonstrates exceptional contiguity with contig N50 of 100.63 Mb and scaffold N50 of 308.03 Mb, substantially outperforming previous Tetrigidae genomes. Repetitive elements constitute 58.79% of the genome, with LINEs being the most abundant class (14.15%). We annotated 17,220 protein-coding genes with functional information successfully assigned to 93.82% of them. This high-quality genomic resource provides an essential foundation for investigating the genetic mechanisms underlying winglessness, ecological specialization, and restricted distribution patterns in Gibbotettix, while also offering valuable insights for biodiversity conservation of these sensitive bioindicator species in their increasingly threatened forest habitats.
Methods
Sample collection and sequencing
G. parvipulvillus individuals were collected from Jiuwan Mountain, Rongshui County, Guangxi, China. For genome sequencing, a total of four adult individuals (All males) were used. Thoracic muscles and other body tissues were carefully dissected and processed for different sequencing applications. High-molecular-weight genomic DNA was isolated from the pooled thoracic muscles using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol.
For PacBio HiFi sequencing, approximately 50 μg of high-quality genomic DNA was used to construct a SMRTbell library according to the manufacturer’s protocol. The library was size-selected to enrich for fragments approximately 15-20 kb in length and sequenced on a PacBio Sequel IIe platform using circular consensus sequencing (CCS) to generate high-fidelity long reads. For complementary short-read sequencing, genomic DNA from the same individual was sheared into shorter fragments (approximately 350 bp), and libraries were prepared following standard protocols. Paired-end sequencing (2 × 150 bp) was performed on a BGISEQ-500 platform to generate high-coverage short reads for error correction and heterozygosity assessment. A Hi-C library was constructed from the same specimen following an in situ Hi-C protocol using the restriction enzyme MboI17. After chromatin cross-linking, digestion, proximity ligation, and reverse cross-linking, the Hi-C library was sequenced on a BGISEQ-500 platform with paired-end reads of 150 bp to enable chromosome-level scaffolding. For transcriptome sequencing, total RNA was extracted from tissues of a single G. parvipulvillus individual to avoid genetic heterogeneity and ensure consistent gene expression profiling. Following quality assessment and poly(A) enrichment, RNA-seq libraries were prepared using random priming and PCR amplification. The libraries were sequenced on a BGISEQ-500 platform with paired-end reads of 150 bp.
All raw sequencing data were quality-filtered using standard bioinformatic pipelines to obtain clean data for subsequent analysis. The specimen was deposited at the Entomological Collection of Guangxi Normal University for long-term preservation and reference, with ID XDTBZ-1. Computational analyses for genome assembly and annotation were performed on a high-performance computing cluster with sufficient storage and processing capabilities.
Genome assembly
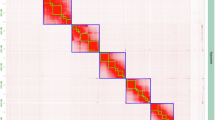
We performed de novo assembly of the G. parvipulvillus genome using a hybrid approach that incorporated 47.43 Gb (42-fold coverage) of PacBio HiFi reads, 47.32 Gb (42-fold coverage) of clean Illumina short reads, and 68.34 Gb (60-fold coverage) of high-throughput chromatin conformation capture (Hi-C) data (Table S1). The PacBio HiFi reads were assembled into contigs using hifiasm v0.19.818 with default parameters, providing a foundation of highly accurate contiguous sequences. For chromosome-level scaffolding, raw Hi-C data were processed using HiC-Pro v3.1.02519 to filter low-quality reads and remove potential artifacts. The clean Hi-C data were subsequently aligned to the assembled contigs using the Juicer pipeline v1.620 to generate a chromosome-scale interaction matrix, which enabled the ordering and orientation of contigs into pseudochromosomes using HapHiC v1.0.521. The resulting assembly was manually curated using Juicebox v2.1722 to ensure accurate chromosome construction based on the Hi-C contacts. Following the clustering, ordering, and orientation of scaffolds to restore their relative locations, the Hi-C contact map validated the robustness and completeness of the genome assembly (Fig. 1). The contact map shows clear diagonal enrichment and distinct chromosomal blocks, confirming proper chromosome assembly and scaffolding.

Hi-C contact map showing chromosome-level assembly validation of the G. parvipulvillus genome. The heatmap displays interaction frequencies between genomic regions, with darker colors indicating higher contact probabilities.
The final assembled genome size was 1.42 Gb, of which 1.39 Gb (representing 97.88% of the assembly) was successfully anchored onto 7 pseudochromosomes (Fig. 2A; Table 1), providing a nearly complete chromosome-level representation of the G. parvipulvillus genome. To identify potential sex chromosomes in the G. parvipulvillus genome, we analyzed sequencing coverage patterns of hifi reads across all pseudochromosomes using mosdepth v0.3.323. Coverage depth analysis revealed six pseudochromosomes (Chr01-Chr06) showed consistent mean coverage depths ranging from 33.34 × to 35.04 × , closely matching the expected whole-genome sequencing depth. In contrast, Chr07 exhibited a significantly reduced mean coverage of 18.04 × , approximately half (51.5%) the average coverage of the other chromosomes. This distinct coverage pattern is characteristic of a heterogametic sex chromosome system, where the heterogametic sex (in this case, likely XO male) possesses only a single copy of the sex chromosome. The approximately 50% reduction in sequencing coverage on Chr07 compared to the autosomes provides strong evidence that Chr07 represents the X chromosome in G. parvipulvillus.

Chromosome-level genome features of G. parvipulvillus. (A) Circos plot displaying the seven pseudochromosomes with length indicated in megabases. (B) Gene density distribution shown as gene count per 100 kb window. (C-I) Distribution of major repetitive element classes across the genome (100 kb overlapping sliding windows): (C) LTR retrotransposons, (D) LINE elements, (E) SINE elements, (F) DNA transposons, (G) Penelope elements, (H) Rolling Circle elements, and (I) unclassified repetitive sequences. (J) GC content variation across the genome shown in 100 kb overlapping sliding windows. The centre image shows the lateral habitus of an adult male G. parvipulvillus.
The assembled pseudochromosomes ranged in size from 103.2 Mb to 417.9 Mb, with a mean length of 139.05 Mb. The assembly demonstrates exceptional contiguity, with a contig N50 of 100.63 Mb and a scaffold N50 of 308.03 Mb. These metrics represent a substantial improvement over previously published Tetrigidae genomes, including Tetrix japonica (contig N50: 322.9 Kb; scaffold N50: 217.25 Mb), Zhengitettix transpicula (contig N50: 52.73 Mb; scaffold N50: 124.72 Mb), and Eucriotettix oculatus (contig N50: 2.09 Mb; scaffold N50: 123.82 Mb) (Table 1). The exceptional contiguity of our assembly is attributable to the combined advantages of high-accuracy HiFi long reads for resolving repetitive regions and Hi-C data for chromosome-scale scaffolding, resulting in one of the most contiguous genomes available for Orthoptera to date. This high-quality chromosome-level assembly provides a robust foundation for subsequent analyses of genome architecture, gene content, and evolutionary patterns in G. parvipulvillus, enabling comprehensive investigations into the genetic basis of its distinctive morphological features and ecological specializations.
Genome annotation
Comprehensive identification of repetitive elements in the G. parvipulvillus genome was conducted using an integrated approach combining both homology-based and de novo methodologies. We employed the EarlGrey pipeline, which leverages multiple repeat identification tools to maximize detection accuracy and sensitivity24. First, a de novo repeat library was constructed using RepeatModeler v2.0.525, followed by classification and extension of the identified repeat families. The resulting custom repeat library was then complemented with known arthropod repeat sequences from the Repbase database v21.1226. Finally, RepeatMasker v4.1.527 was used to systematically identify and mask repetitive elements throughout the genome assembly.
This integrated approach revealed that repetitive elements comprise a substantial portion of the G. parvipulvillus genome, totaling 836.96 Mb and accounting for 58.79% of the total genome assembly (Fig. 2C-I, Table S2). Among the identified repeats, LTR retrotransposons occupied 106.69 Mb (7.49%, Fig. 2C), while LINE elements were the most abundant class at 201.42 Mb (14.15% of the genome, Fig. 2D). SINE elements contributed a minor portion at 0.74 Mb (0.05%, Fig. 2E). DNA transposons occupied 84.25 Mb (5.92%, Fig. 2F), and Penelope elements accounted for 41.35 Mb (2.90%, Fig. 2G). Rolling Circle elements represented 8.01 Mb (0.56%, Fig. 2H). Other repetitive sequences, including simple repeats, microsatellites, and non-coding RNA genes, collectively accounted for 75.24 Mb (5.28%). Notably, a significant proportion of repetitive elements (337.46 Mb, 23.70%) could not be classified into established categories, suggesting potential lineage-specific repeat innovations in the G. parvipulvillus genome (Fig. 2I).
Following repeat masking, protein-coding gene annotation was performed using a comprehensive pipeline that integrated evidence from three complementary approaches: homology-based prediction, transcriptome-aligned evidence, and ab initio gene modeling. For homology-based annotation, we used protein sequences from closely related Tetrigidae species, including Zhengitettix transpicula and Eucriotettix oculatus (Table 1), which were aligned to the G. parvipulvillus genome using GeneWise v2.4.128. Transcriptome-based evidence was generated by mapping RNA-seq reads to the genome using HISAT2 v2.2.129, which provided valuable information for identifying exon-intron boundaries and alternatively spliced isoforms. Concurrently, ab initio gene prediction was conducted with AUGUSTUS v3.5.030, which was specifically trained using the G. parvipulvillus transcriptome data to improve prediction accuracy for this species.
Homology-based prediction identified 19,508 protein-coding genes using closely related Tetrigidae protein sequences. Transcriptome-based evidence mapping resulted in 16,909 genes with strong RNA-seq support, providing high-confidence annotations for actively expressed loci. The ab initio AUGUSTUS prediction, trained specifically on G. parvipulvillus transcriptome data with an accuracy of 66.36%, predicted 50,432 potential protein-coding genes. The predictions from all three approaches were subsequently integrated using the GETA pipeline (https://github.com/chenlianfu/geta) to generate a comprehensive and non-redundant set of protein-coding gene models. This integrated approach resulted in the identification of 17,220 protein-coding genes in the G. parvipulvillus genome (Fig. 2B, Table 1). Functional annotation of the predicted gene models was conducted through sequence similarity searches against the NCBI non-redundant protein database (Nr) and protein domain identification using InterProScan with parameters “-appl Pfam,SUPERFAMILY--goterms--iprlookup”. This comprehensive functional annotation process successfully assigned putative functions to 16,157 genes, representing 93.82% of the total predicted gene set (Table S3). To assess the completeness of the gene annotation, we performed a BUSCO analysis on these predicted proteins against the Insecta lineage dataset (insecta_odb12), which demonstrated an exceptionally high level of completeness with 94.7% complete and 2.1% fragmented BUSCO genes, indicating the high quality of the gene prediction31. The high proportion of functionally annotated genes further validates the quality of our gene prediction and provides a valuable resource for future comparative and functional genomic studies in Tetrigidae and other orthopteran insects.
Data Records
The chromosome-level genome assembly of G. parvipulvillus has been deposited in the National Center for Biotechnology Information (NCBI) GenBank database under the accession number JBNFPV00000000032. All raw sequencing data generated in this study have been made publicly available through the NCBI Sequence Read Archive (SRA) under the accession number SRP58027033. The PacBio HiFi long-read sequencing data used for primary contig assembly are accessible under accession number SRR33238104. Illumina short-read whole genome sequencing data used for error correction and heterozygosity assessment are available under accession number SRR33238103. The Hi-C sequencing data utilized for chromosome-level scaffolding can be accessed under accession number SRR33238101. Additionally, the RNA-sequencing data used for genome annotation and transcriptome analysis have been deposited under accession number SRR33238102.
Technical Validation
We employed both sequence alignment metrics and established benchmarking tools to ensure the assembly met high-quality standards for accuracy, completeness, and contiguity. Read mapping analysis demonstrated exceptional concordance between the assembly and the original sequencing data. Using BWA v0.7.1734, we aligned the Illumina short reads to the genome assembly, achieving a mapping rate of 98.54%. Similarly, 99.35% of the PacBio HiFi reads successfully mapped to the assembly, confirming excellent representation of the input sequencing data. For transcriptome validation, RNA-seq reads were aligned to the genome using HISAT2 v2.2.129, yielding a mapping rate of 98.09%, which indicates comprehensive representation of transcribed regions within the assembly. Base-level accuracy of the assembly was evaluated using Merqury35, which leverages k-mer profiles from the sequencing data to assess assembly quality. The genome exhibited a remarkably high covered rate of 98.91%, with only 1.80% of the assembly designated as low-confidence regions. The read-level assembly quality indicator (R-AQI) of 99.07 and scaffold-level assembly quality indicator (S-AQI) of 98.53 further confirm the exceptional accuracy of the assembly. The number of ambiguous bases (N) in the entire genome was exceptionally low at only 5,137 (approximately 360 N’s per gigabase), demonstrating the completeness of the sequence data. To assess gene space completeness, we performed Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis using BUSCO v5.8.231 against the insecta_odb12 database. The assembly contained 98.8% of the core orthologous genes (1367 BUSCO groups), with 1325 single-copy (96.9%), 26 duplicated (1.9%), 9 fragmented (0.7%), and 7 missing (0.5%) BUSCO genes identified. This BUSCO completeness score exceeds those reported for the three previously sequenced Tetrigidae congeners (Table 1), confirming the exceptional quality of our gene space representation. The high-quality metrics across multiple validation approaches—including read mapping rates, base-level accuracy measurements, minimal sequence gaps, and comprehensive gene space representation—collectively demonstrate that our G. parvipulvillus genome assembly provides an exceptionally complete and accurate reference for future genomic studies.
Code availability
While no complex custom scripts exceeding standard bioinformatics practices were developed, all data processing and filtering steps have been documented with specific command-line instructions. All bioinformatics workflows were implemented using established, publicly available software tools with comprehensive documentation. The workflow adheres to existing dependency and workflow management standards, utilizing conda for dependency management and containerized environments to ensure consistent software execution across different computing platforms. The version and code/parameters of software have been detailed described in Methods.
References
Deng, W. A., Zheng, Z. M. & Wei, S. Z. Two new species of the genus Gibbotettix Zheng, 1992 (Orthoptera: Tetrigidae, Cladonotinae) from China. Zootaxa 4200, 426–436, https://doi.org/10.11646/zootaxa.4200.3.8 (2016).
Guerra, P. A. Evaluating the life-history trade-off between dispersal capability and reproduction in wing dimorphic insects: a meta-analysis. Biological Reviews 86, 813–835, https://doi.org/10.1111/j.1469-185X.2010.00172.x (2011).
Zha, L. S., Wen, T. C., Yu, F. M. & Hyde, K. D. A taxonomic review of the genus Gibbotettix with description of one new species (Orthoptera: Tetrigidae). Journal of Natural History 50, 2389–2397, https://doi.org/10.1080/00222933.2016.1193651 (2016).
Kuravová, K., Sipos, J. & Kocárek, P. Energy balance of food in a detrito-bryophagous groundhopper (Orthoptera: Tetrigidae). Peerj 8, https://doi.org/10.7717/peerj.9603 (2020).
Liu, T., Liu, H. Y., Wang, Y. J. & Yang, Y. X. Climate Change Impacts on the Potential Distribution Pattern of Osphya (Coleoptera: Melandryidae), an Old but Small Beetle Group Distributed in the Northern Hemisphere. Insects 14, https://doi.org/10.3390/insects14050476 (2023).
Jiang, Y. L. et al. Similar pattern, different paths: tracing the biogeographical history of Megaloptera (Insecta: Neuropterida) using mitochondrial phylogenomics. Cladistics 38, 374–391, https://doi.org/10.1111/cla.12494 (2022).
Valero, K. C. W. et al. Patterns, Mechanisms and Genetics of Speciation in Reptiles and Amphibians. Genes 10, https://doi.org/10.3390/genes10090646 (2019).
Li, R., Qin, Y. C., Rong, W. T., Deng, W. A. & Li, X. D. Chromosome-level genome assembly of the pygmy grasshopper Eucriotettix oculatus (Orthoptera: Tetrigoidea). Scientific Data 11, https://doi.org/10.1038/s41597-024-03276-2 (2024).
Guan, D. L., Chen, Y. Z., Qin, Y. C., Li, X. D. & Deng, W. A. Chromosomal-Level Reference Genome for the Chinese Endemic Pygmy Grasshopper, Zhengitettix transpicula, Sheds Light on Tetrigidae Evolution and Advancing Conservation Efforts. Insects 15, https://doi.org/10.3390/insects15040223 (2024).
Zhang, R. J., Zhao, C. L., Wu, F. P. & Deng, W. A. Molecular data provide new insights into the phylogeny of Cladonotinae (Orthoptera: Tetrigoidea) from China with the description of a new genus and species. Zootaxa 4809, 547–559, https://doi.org/10.11646/zootaxa.4809.3.8 (2020).
Li, W. C., Boonmee, S., Eungwanichayapant, P. D. & Zha, L. S. Notes on Gibbotettix and Austrohancockia (Tetrigidae: Cladonotinae) with Description of Two New Species. Entomological News 127, 303–314, https://doi.org/10.3157/021.127.0401 (2018).
Deng, W. A. Taxonomic revision of the subfamily Cladonotinae (Orthoptera: Tetrigidae) from China with description of three new species. Zootaxa 4789, 403–440, https://doi.org/10.11646/zootaxa.4789.2.4 (2020).
Qiu, Z. Y., Liu, F., Lu, H. M. & Huang, Y. Characterization and analysis of a de novo transcriptome from the pygmy grasshopper Tetrix japonica. Molecular Ecology Resources 17, 381–392, https://doi.org/10.1111/1755-0998.12553 (2017).
Liu, Y. X., Li, X. J. & Lin, L. L. Transcriptome of the pygmy grasshopper Formosatettix qinlingensis (Orthoptera: Tetrigidae). Peerj 11, https://doi.org/10.7717/peerj.15123 (2023).
Ren, L. P. et al. Chromosome-level de novo genome assembly of Sarcophaga peregrina provides insights into the evolutionary adaptation of flesh flies. Molecular Ecology Resources 21, 251–262, https://doi.org/10.1111/1755-0998.13246 (2021).
Lee, J. et al. A chromosome-level genome assembly of wild silkmoth, Bombyx mandarina. Scientific Data 12, https://doi.org/10.1038/s41597-025-04395-0 (2025).
Cardamone, F., Zhan, Y., Iovino, N. & Zenk, F. Chromosome Conformation Capture Followed by Genome-Wide Sequencing (Hi-C) in Drosophila Embryos. Methods in molecular biology (Clifton, N.J.) 2655, 41–55, https://doi.org/10.1007/978-1-0716-3143-0_4 (2023).
Cheng, H. Y., Concepcion, G. T., Feng, X. W., Zhang, H. W. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–+, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology 16, https://doi.org/10.1186/s13059-015-0831-x (2015).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Systems 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Zeng, X. F. et al. Chromosome-level scaffolding of haplotype-resolved assemblies using Hi-C data without reference genomes. Nature Plants 10, https://doi.org/10.1038/s41477-024-01755-3 (2024).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Systems 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Pedersen, B. S. & Quinlan, A. R. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics (Oxford, England) 34, 867–868, https://doi.org/10.1093/bioinformatics/btx699 (2018).
Baril, T., Galbraith, J. & Hayward, A. Earl Grey: A Fully Automated User-Friendly Transposable Element Annotation and Analysis Pipeline. Molecular Biology and Evolution 41, https://doi.org/10.1093/molbev/msae068 (2024).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences of the United States of America 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Bao, W. D., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics Chapter 4, Unit 4.10, https://doi.org/10.1002/0471250953.bi0410s05 (2004).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome research 14, 988–995, https://doi.org/10.1101/gr.1865504 (2004).
Kim, D., Landmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–U121, https://doi.org/10.1038/nmeth.3317 (2015).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic acids research 34, W435–439, https://doi.org/10.1093/nar/gkl200 (2006).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods in Molecular Biology 1962, 227–245, https://doi.org/10.1007/978-1-4939-9173-0_14 (2019).
Guan, D.-L. Genbank https://identifiers.org/ncbi/insdc.gca:GCA_050613505.1 (2025).
Guan, D.-L. NCBI. Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP580270 (2025).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595, https://doi.org/10.1093/bioinformatics/btp698 (2010).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Acknowledgements
The project is supported by the Science & Technology Fundamental Resources Investigation Program of China (2023FY100200), the National Natural Science Foundation of China (32360124).
Author information
Authors and Affiliations
Contributions
W.-A.D. conceived the project and supervised this study. W.-A.D., B.-W.Z. and C.-L.T. performed the experiments and collected samples. C.-L.T. conducted data analysis. B.-W.Z. and D.-L.G. performed computational analysis. W.-A.D. wrote the manuscript. D.-L.G revised the manuscript. All authors read and approved the final manuscript and all authors commented on the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41597_2025_5474_MOESM2_ESM.xlsx
Table S2. Classification and statistics of repetitive sequences identified in the Gibbotettix parvipulvillus genome assembly at the superfamily level.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Deng, WA., Zheng, BW., Teng, CL. et al. Chromosome-level genome assembly and annotation of the pygmy grasshopper Gibbotettix parvipulvillus. Sci Data 12, 1119 (2025). https://doi.org/10.1038/s41597-025-05474-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05474-y
This article is cited by
-
Evolutionary dynamics of repetitive elements and genome size in Tetrigidae (Orthoptera: Caelifera)
Scientific Reports (2025)
