
Abstract
Humphead wrasse, Cheilinus undulatus, is an endangered fish species with high economic and ecological value as well as natural sex change from female to male, while sexual selection occurs in breeding aggregations. In our present study, we constructed the first gap-free telomere-to-telomere (T2T) genome assembly for humphead wrasse, by integration of PacBio HiFi, ONT Ultra-long and Hi-C sequencing techniques. With 99% of the entire sequences anchored into 24 chromosomes, this haplotypic genome assembly spans approximately 1.25 Gb and presents a complete set of 48 telomeres and 24 centromeres. In terms of correctness (quality value QV: 53.447) and completeness (BUSCO score: 99.3%), this chromosome-scale assembly is indeed of high quality. We predicted 658.03 Mb of repetitive sequences and annotated 26,609 protein-coding genes in the assembled genome. This high-quality T2T genome assembly not only facilitates the genetic conservation of humphead wrasse, but also offers fundamental genomic data for supporting in-depth investigations on functional genomics, genetic diversity, and selective breeding for this economically important teleost.
Similar content being viewed by others


Background & Summary
Humphead wrasse (Cheilinus undulates), also commonly known as Maori or Napoleon wrasse, is an endangered fish species with significant ecological importance for coral reef ecosystems. As a member of the Labridae family within the order Perciformes, it is characterized by large size and striking appearance1,2. Moreover, it has a sparse population, slow growth, and a long lifespan (over 30 years), as well as intricate reproductive behaviors, but it significantly contributes to bioerosion and sand production3. Previous investigations indicate that its populations are declining alarmingly because of ongoing overharvesting, habitat degradation, and climate change impacts4,5. Humphead wrasse has been classified as ‘Endangered’ on the IUCN (International Union for Conservation of Nature) Red List and is included in the CITES (Convention on International Trade in Endangered Species of Wild Fauna and Flora) Appendix II6.
Like many other environmentally-sensitive sex-changing species, humphead wrasse exhibits protogynous hermaphroditism, i.e., transitioning from female to male at approximately 8-9 years old, after attaining female sexual maturity at 5–7 years7,8. Males usually follow two distinct developmental pathways to achieve diandry, either developing directly from juveniles into small males (smaller than the smallest mature female) or transitioning from adult females through sex change to become large males (exceeding female size)4. However, the detailed molecular mechanisms of its sex change remain largely unknown. Additionally, due to its unique visual system and fused pharyngeal bones, it always serves as an excellent model organism for studying opsin evolution in coral reef fishes, and for comparative studies with other fish genomes to demonstrate specific opsin gene expansions in humphead wrasse9.
In a previous genome study for humphead wrasse, a draft chromosome-level genome assembly was reported9. Nevertheless, this assembly version contains excessive gaps accompanied by low BUSCO values, leading to significant fragment loss that impairs both genome completeness and annotation accuracy. Here, utilizing cutting-edge high-throughput sequencing platforms including PacBio HiFi and Oxford Nanopore Technologies (ONT) ultra-long technology, we produced a refined telomere-to-telomere (T2T) chromosome-level genome assembly for humphead wrasse. This improved assembly demonstrates superior scaffold N50 and BUSCO scores, as well as gap-free genome sequence with encouraging details of telomeres and centromeres. This new genome assembly not only provides a valuable genetic resource for in-depth investigations on population genetics and conservation biology of humphead wrasse, but also supports comparative and molecular studies on the regulation of natural sex change and opsin evolution in various vertebrates.
Methods
Sample collection
We obtained an adult humphead wrasse (Fig. 1a) from Guangdong Marine Fisheries Experimental Centre, an offsite facility of the Agro-Tech Extension Center of Guangdong Province, which is situated in Huizhou city, Guangdong province, China. Muscle tissue was collected for whole-genome sequencing, and ten tissues (including intestine, spleen, lung, heart, liver, muscle, gill, eye, skin, and gonad) were sampled for transcriptome sequencing. The sampling procedure and experimental workflow were performed in accordance with the guidelines and approval from the Animal Ethics Committee of Shenzhen University (Shenzhen, China).

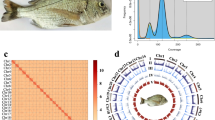
A T2T genome assembly of the humphead wrasse. (a) An image of the sequenced fish. (b) A GenomeScope k-mer plot. (c) The Hi-C contact matrices contain 24 unique blocks in total. (d) A Circos plot displaying the primary genome characteristics, with features annotated from the outermost to innermost rings, (I) the 24 chromosomes, (II) gene density, (III) GC skew, (IV) GC content, (V) tandem repeats, and (VI) transposons.
DNA extraction and genome sequencing
Genomic DNA (gDNA) was extracted from the muscle tissue using a modified CTAB method10. The extracted gDNA was used for construction of a BGISeq DNA PCRfree library, which was then sequenced on a BGI T7 platform (MGI, Shenzhen, China). A total of 57.98 Gb of raw reads (150 bp in length) were generated, among them low-quality reads and adaptor sequences were filtered using Trimmomatic (v0.40)11 with default settings. Finally, we obtained 56.64 Gb of clean reads for estimating genome size and assembling sequences.
Moreover, we prepared long-read libraries using the PacBio Sequel II System and SMRTbell Express Template Prep Kit 3.0 (Pacific Biosciences, Menlo Park, CA, USA) for HiFi sequencing. The CCS software (SMRT Link v9.0)12 was then applied to generate consensus sequences. In this study we yielded approximately 113.26 Gb of consensus reads, with an average length of 19.07 kb.
ONT technology was applied by construction of an ultra-long library and then sequencing of one flow cell on a PromethION platform (Oxford Nanopore Technologies Co., UK). The raw reads were first refined to remove those with quality value (QV) below 7. Subsequently, Porechop (https://github.com/rrwick/Porechop) was applied to eliminate adaptors, and Filtlong (https://github.com/rrwick/Filtlong) was employed to remove those reads shorter than 30 kb and mean read quality scores less than 90%. Finally, we obtained a total of 27.88 Gb clean reads, with an average read length of 96.15 kb and an N50 length of 100 kb.
DNA libraries for Hi-C sequencing were constructed with a GrandOmics Hi-C kit (GrandOmics, China), employing DpnII as the restriction enzyme, in accordance with the manufacturer’s instructions. By using the Illumina Novaseq system (Illumina Inc., San Diego, CA, USA), we produced 74.82 Gb of raw reads from the Hi-C libraries. We then employed Trimmomatic (v0.4)11 to remove low-quality reads (quality scores <20), adapter sequences, and reads shorter than 36 bp. After filtering, 71.53 Gb of clean data were available for subsequent chromosome scaffolding.
RNA extraction and transcriptome sequencing
We used poly‐T oligo‐attached magnetic beads to purify mRNAs from the lung, heart, liver, muscle, gill, eye, skin, and gonad tissues. Sequencing libraries were generated from the purified mRNAs using the VAHTS Universal V6 RNA-seq Library Kit for MGI (Vazyme, Nanjing, China) following the manufacturer’s recommendations with unique index codes. Library quantification and size were assessed using Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Subsequently, sequencing was performed on a MGI-SEQ. 2000 platform by Frasergen Bioinformatics Co. Ltd. (Wuhan, China).
To obtain clean reads, adaptor sequences and low-quality raw reads were filtered via SOAPfilter (v2.2)13 with default parameters. In the end, the clean reads of the ten tissues (namely the intestine, spleen, lung, heart, liver, muscle, gill, eye, skin, and gonad) were 5.77, 5.58, 6.29, 8.56, 5.64, 8.38, 6.10, 6.14, 5.96 and 13.58 Gb, respectively. These retained data were collected for annotation of gene structures.
Genome assembly
Genome-size estimation
We employed Jellyfish (v2.2.6)14 and GenomeScope (v2.0)15 to analyze the K-mer frequency distribution of the BGI clean reads. Our results showed that the humphead wrasse genome was estimated to be 1.17 Gb in length, with a genomic heterozygosity rate of 0.27% (Fig. 1b).
De novo genome assembly
We applied HiFiasm (v0.19.5)16 to assemble HiFi and ONT long reads into contigs, which were then polished using T2T-polish17 with the optimized parameter set to task = best using the BGI short reads. The initial genome assembly had a total length of 1.253 Gb, with a contig N50 of 54.5 Mb and an organized of 53 contigs.
Construction of chromosomes and gap filling
Hi-C reads were aligned to the primary genome assembly using Bowtie2 (v2.3.2)18, followed by identification of valid contact paired reads through the HiC-Pro (v2.8.1) pipeline19. The assembled contigs were anchored to chromosomes using these Hi-C valid reads through the 3D-DNA pipeline20 with the parameter -r 0, followed by manual refinement of the chromosome-level scaffolds in JuiceBox21. To close nucleotide gaps in the chromosome-level genome assembly, we utilized TGS-GapCloser (v1.1.1)22 with default parameters, leveraging both HiFi and ONT long reads. The final genome assembly spans 1.25 Gb, with 99% of the primary sequences anchored to 24 chromosomes, achieving a contig N50 of 54.51 Mb (Fig. 1c,d and Table 1).
Identification of centromere and telomere sequences
Telomere sequences were identified by detecting (TTAGGG/CCCTAA) repeats in telomeric regions, while centromeres were localized using the Centromics program (https://github.com/ShuaiNIEgithub/Centromics) to analyze HiFi sequencing data, Hi-C data, and the final genome assembly. Finally, we revealed that the humphead wrasse chromosomes possessed 48 telomeres and 24 centromeres (see more details in Fig. 2 and Table 1).

An overview of the T2T gap-free genome assembly of humphead wrasse. The telomere regions are shown as orange segments at both ends of each chromosome, while the centromere region appears as a gully area within the chromosome.
Genome annotation
Repeat annotation. Tandem repeats were identified using Tandem Repeats Finder (TRF, v4.09.1)23 with the following parameters: 2 7 7 80 10 50 2000. Moreover, transposable elements (TEs) were detected through an integration of de novo prediction and homology searches at both DNA and protein levels. LTR retrotransposons were initially identified using LTR_FINDER (v1.0.7)24 at the DNA level, while RepeatModeler (v2.0.1)25 generated a classified de novo repeat library. Subsequently, RepeatMasker (v4.1.2)26 performed comparative analyses against both the Repbase TE database27 and the newly constructed repeat library. Protein-level TE annotation was performed using RepeatProteinMask26 against the transposable element protein database. A total of 658.03 Mb repetitive sequences were detected in the humphead wrasse genome assembly (Table 2).
Gene annotation
Protein-coding genes were predicted using a combination of homology-based, ab initio and transcriptome-assisted annotation approaches. The homology-based annotation was initiated by performing Tblastn (v2.11.0+)28 searches against our assembly using protein sequences from four representative species, including yellowfin seabream (Acanthopagrus latus), sharksucker (Echeneis naucrates), zebrafish (Danio rerio), and medaka (Oryzias latipes). The high-quality alignments were subsequently refined using Exonerate (v2.4.0)29 for precise gene model prediction. The de novo annotation was performed using Augustus (v3.4.0)30 and GlimmerHMM (v3.0.4)31. For the transcriptome-assisted annotation, RNA-seq reads were first aligned to the reference genome using HiSat2 (v2.2.1)32, followed by transcript assembly through a genome-guided approach implemented in StringTie (v2.1.7)33. Predicted gene models were integrated and refined using MAKER (v3.01.03)34 to generate a non-redundant gene set. Final annotation improvements, including UTR annotation and alternative splicing variant prediction, were accomplished through the PASA pipeline (v2.4.1)35. Ultimately, we annotated a total of 25,064 protein-coding genes, with an average gene length of 27.84 kb and a mean coding sequence (CDS) size of 1,745.64 bp (see Table 3).
Functional annotations
Functional annotation was performed by aligning protein sequences against multiple databases (NCBI NR, KEGG36, GO37, TrEMBL and Swiss-Prot38) using DIAMOND BLASTP (v2.0.7)39, with assignments based on best matches. Functional annotations were assigned to 24,789 genes (98.90%) with supportive evidence from at least one database (see more details in Table 3).
Annotation of non-coding RNA genes
tRNAscan-SE (v2.0.9)40 with default settings was utilized to detect tRNA genes. Moreover, we applied RNAmmer (v1.2)41 to identify rRNA sequences. Annotation of MiRNA and snRNA genes was performed using Infernal (v1.1.2)42 through homology searches against the Rfam database (v14.6)43. Finally, a total of 2,221 rRNAs, 3,020 tRNAs, 781 miRNAs, and 646 snRNAs were predicted (see Table 4 for more details).
Data Records
All genomic data are publically available from China National GeneBank DataBase (CNGBdb) under the project ID no. CRA02360944. The genome assembly has been submitted to the GenBank database with the accession number JBMUSF01000000045. In addition, comprehensive documentation regarding the genome assembly, gene structures, functional annotations, and repeat elements of humphead wrasse has been deposited on Figshare46.
Technical Validation
Evaluation of the genome assembly
Genome completeness was assessed using BUSCO (v5.2.2)47 against the actinopterygii_odb10 database (3,640 single-copy orthologs). Our results demonstrated 98.9% complete gene coverage (including 98.4% single-copy and 0.5% duplicated genes), with only 0.4% fragmented sequences (see Table 5). Moreover, the Merqury (v1.3)48 analysis estimated a genome assembly quality value of 53.45. Genome assembly accuracy was evaluated by aligning sequencing datasets, revealing mapping rates of 96.58% (RNA-Seq reads), 99.64% (for BGI reads), 99.93% (for PacBio reads), and 100% (for ONT reads). These analyses collectively validate the high-quality of this humphead wrasse genome assembly.3
Collinearity analysis
GenomeSyn (v1.2.7)49 was employed for whole-genome synteny comparison between the newly assembled genome and the previously published version (GCF_018320785.1)9. Our findings revealed good one-to-one chromosomal synteny between both assemblies (Fig. 3), which further validates that our present assembly of the humphead wrasse genome is indeed of high quality.

Good synteny of chromosomes between the newly assembled genome and the previously published version9.
Code availability
This study did not employ any custom code. In cases where specific parameters were unavailable for any software type, the default settings recommended by the developers were applied.
References
Oktaviani, D. et al. Initiating Napoleon wrasse (Cheilinus undulatus Ruppell, 1835) as watching species object in Banda Islands marine ecotourism. IOP Conference Series: Earth and Environmental Science 800, 012053 (2021).
Salvador, M. L. et al. Intact shallow and mesophotic assemblages of large carnivorous reef fishes underscore the importance of large and remote protected areas in the Coral Triangle. Aquatic Conserv: Mar Freshw Ecosyst 34, e4108 (2024).
Friedlander, A. M. et al. Assessing and managing charismatic marine megafauna in Palau: Bumphead parrotfish (Bolbometopon muricatum) and Napoleon wrasse (Cheilinus undulatus). Aquatic Conserv: Mar Freshw Ecosyst 33, 349–365 (2023).
Sadovy, Y. et al. The humphead wrasse, Cheilinus undulatus: Synopsis of a threatened and poorly known giant coral reef fish. Rev Fish Biol Fisher 13, 327–364 (2003).
Russell, B. Cheilinus undulatus. The IUCN Red List of Threatened Species 2004, e.T4592A11023949 (2004).
Donaldson, T. J. & Sadovy, Y. Threatened fishes of the world: Cheilinus undulatus Rüppell, 1835 (Labridae). Environ Biol Fish 62, 428 (2001).
Sadovy de Mitcheson, Y., Liu, M. & Suharti, S. Gonadal development in a giant threatened reef fish, the humphead wrasse Cheilinus undulatus, and its relationship to international trade. J Fish Biol. 77, 706–718 (2010).
Ji, X. et al. Identification of SF-1 and FOXL2 and their effect on activating P450 aromatase transcription via specific binding to the promoter motifs in sex reversing Cheilinus undulatus. Front Endocrinol 13, 863360 (2022).
Liu, D. et al. Chromosome‐level genome assembly of the endangered humphead wrasse Cheilinus undulatus: Insight into the expansion of opsin genes in fishes. Mol Ecol Resour 21, 2388–2406 (2021).
Gelvin, S. B., Schilperoort R. A. Plant molecular biology manual. Springer (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Rhoads, A. & Au, K. F. PacBio sequencing and its applications. Genom. Proteom. Bioinfor. 13, 278–289 (2015).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with HiFiasm. Nat. Methods 18, 170–175 (2021).
Cartney, A. M. et al. Chasing perfection: validation and polishing strategies for telomere-to-telomere genome assemblies. Nat Methods 19, 687–695 (2022).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Xu, M. et al. TGS-GapCloser: fast and accurately passing through the Bermuda in large genome using error-prone third-generation long reads. BioRxiv 831248 (2019).
Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Xu, Z. & Wang, H. LTR-FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, 265–268 (2007).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. PNAS 117, 9451–9457 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinforma. Chapter 4, 4.10.1–4.10.14 (2009).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. Dna 6, 1–6 (2015).
Gertz, E. M., Yu, Y. K., Agarwala, R., Schäffer, A. A. & Altschul, S. F. Composition-based statistics and translated nucleotide searches: Improving the TBLASTN module of BLAST. BMC Biol. 4, 41 (2006).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user defined constraints. Nucleic Acids Res. 33, W465–W467 (2005).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan. & GlimmerHMM: two open-source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Kim, D. et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 37, 907–915 (2019).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Cantarel, B. L. et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, 1–22 (2008).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114 (2012).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 (2000).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 49, D192–D200 (2021).
NCDC GSA https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA036754 (2025).
Zhang, K. et al. Cheilinus undulatus isolate JC-2025a, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JBMUSF010000000 (2025).
Zhang, K. et al. The genome annotation files of Cheilinus undulatus. figshare https://doi.org/10.6084/m9.figshare.28887965 (2025).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Zhou, Z. et al. GenomeSyn: a bioinformatics tool for visualizing genome synteny and structural variations. Journal of genetics and genomics 49, 1174–1176 (2022).
Acknowledgements
This project was supported by Research on breeding technology of candidate species for Guangdong modern marine ranching (No. 2024-MRB-00-001), Guangdong Basic and Applied Basic Research Foundation (No. 2023A1515110554), Shenzhen Science and Technology Program (No. 827-0001055), and Research Initiation Fund for Young Faculty Members at Shenzhen University (No. 000001032214).
Author information
Authors and Affiliations
Contributions
Q.S. and J.W. conceived this study. K.Z. performed data analysis; J.C., S.Y. and Y.Z. participated in the collection of samples; J.C., B.D., C.Y. and W.Z. provided research advice; K.Z. wrote the draft manuscript. Q.S. and J.W. revised the manuscript. All authors have read and approved the final manuscript for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, K., Chen, J., Duan, B. et al. A telomere-to-telomere gap-free genome assembly of the endangered humphead wrasse (Cheilinus undulatus). Sci Data 12, 1194 (2025). https://doi.org/10.1038/s41597-025-05475-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05475-x
