
Abstract
Transcription readthrough occurs when RNA polymerase bypasses canonical termination sites, producing elongated RNA molecules called readthrough (RT) transcripts or downstream of gene (DoG) transcripts. Although RT transcripts have been implicated in stress responses and pathological states, their roles in healthy human tissues are poorly understood. This study collected and analyzed RT events across 43 healthy human tissues, identifying 75,248 RT events from 35,720 transcripts across 11,692 genes. The dataset encompasses the sequences, locations, expression profiles, and comprehensive annotation information of corresponding genes for RT transcripts. It provides a thorough elucidation of RT transcriptomics and its significance in gene regulation, offering a wealth of benchmark data to facilitate further research on RT transcripts.
Similar content being viewed by others


Background & Summary
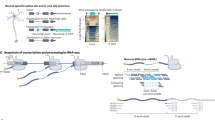
Transcription termination is a critical regulatory step in gene expression, wherein RNA polymerase ceases RNA synthesis and dissociates from the DNA template upon completing transcription1. However, under specific conditions, the transcription machinery may fail to recognize termination signals, resulting in transcription extending beyond the defined gene boundaries—a phenomenon termed transcription readthrough (TRT)2,3,4,5. This process generates elongated RNA molecules known as readthrough (RT) transcripts or downstream of gene (DoG) transcripts, which have been observed under various stress conditions6, including hyperosmotic stress, heat shock7, oxidative stress1,2, hypoxia8,9, viral infections5,10,11, and cancer4,12,13,14,15. These transcripts have been implicated in maintaining chromatin structure and potentially modulating gene expression2,3,16. Furthermore, studies have shown that the three-dimensional organization of chromatin not only affects the initiation and extension of transcription but also plays a significant role in transcription termination. Changes in chromatin structure can influence the behavior of RNA polymerase II, thereby regulating the efficiency and precision of transcription termination17,18.
TRT disrupts gene regulatory networks by extending into downstream genomic regions, potentially triggering unintended transcription of neighboring genes without promoter activation19. This process can also result in functional antisense RNAs that repress gene expression or in the formation of circular RNAs from downstream exons4. Furthermore, RT events may produce RNA chimeras through intergenic exon splicing, some of which are linked to tumor proliferation and cancer survival20. These findings underscore the multifaceted roles of TRT in both normal cellular processes and disease pathogenesis1,4,8,9,21.
Recent evidence demonstrates that RT transcripts are not confined to stress responses or pathological conditions but are also prevalent in healthy human tissues22. This widespread occurrence suggests that RT transcripts may play vital physiological roles in maintaining cellular homeostasis22. However, systematic large-scale investigations into TRT differences between healthy and diseased tissues remain limited. Previous research has primarily targeted cancer-specific chimeric RNAs and the development of specialized disease-focused databases21,23,24, the broader biological implications of RT transcripts, beyond chimeric RNA formation, remain insufficiently explored. Moreover, the current state of TRT research is characterized by fragmented and inconsistent data, with no dedicated platform to support systematic analyses. To address this gap, we generate the comprehensive dataset for TRT in healthy human tissues and developed the online platform (hhrtBase, http://www.hhrtbase.com/). This platform serves as a comprehensive reference, offering an integrated platform for browsing, downloading, and analyzing RT data across various samples. By enabling systematic comparisons, hhrtBase aims to elucidate the functional significance of transcription RT in normal physiology and disease, advancing its applications in biomedical research.
The dataset presented in this study, comprising 75,248 TRT events from 11,692 genes in 43 healthy human tissues. By offering a systematic catalog of TRT events, the dataset enables researchers to investigate the prevalence, distribution, and functional implications of RT transcripts in normal physiology. For example, researchers can use this dataset to identify tissue-specific TRT patterns, correlate TRT events with gene expression profiles, or explore associations between RT transcripts and chromatin organization. Such analyses could reveal novel regulatory mechanisms underlying cellular homeostasis or identify potential biomarkers for physiological states. The dataset’s utility extends to comparative studies between healthy and diseased tissues. For instance, researchers can compare this dataset with cancer TRT data to identify aberrant RT events associated with oncogenesis or tumor progression. Additionally, the dataset supports studies on the evolutionary conservation of TRT events, the impact of genetic variants on RT propensity, and the role of RT transcripts in shaping the non-coding RNA landscape. By providing a robust, curated reference of TRT events in healthy human tissues, this dataset serves as a foundational resource for hypothesis-driven research, enabling scientists to address fundamental questions in gene regulation, chromatin dynamics, and disease biology.
Data Summary
Analysis of 2,759 RNA-seq samples from 43 tissues revealed 75,248 RT events derived from 11,692 genes. The lengths of these RT transcripts varied significantly, ranging from 2,001 base pairs to over 177,501 kilobases (kb) beyond the annotated gene boundaries, with a median length of approximately 7.7 kb. Notably, some RT events exhibited extraordinary extensions exceeding 177 kb (ENSG00000256499), particularly in tissues such as the artery (aorta) and artery (tibial).
The distribution of RT transcripts across 43 tissues revealed variability (Fig. 1). Testis exhibited the highest numbers of RT transcripts, with 3,012 transcripts. Other tissues, including the thyroid, stomach, spleen, prostate, placenta, pituitary, lymph node, lung, gall bladder, endometrium, brain (cerebellum), bone marrow, and appendix, demonstrated moderately elevated RT transcript counts, ranging between 2,000 and 3,000. Most tissues exhibited RT transcripts ranging from 1,000 to 2,000. Conversely, tissues like the tonsil, smooth muscle, rectum, pancreas, and heart (left ventricle) displayed fewer than 1,000 RT transcripts. These differences reflect variations in data volume across tissues. However, whether they indicate actual differences in TRT among tissues requires further in-depth analysis by researchers, including examination of individual samples and expression profiles.

Tissue distribution of RT transcripts in 43 tissues. The first bar chart illustrates the total number of readthrough (RT) transcripts identified in each of the 43 human tissues analyzed. Tissues are categorized based on the number of RT transcripts: >3,000 (red), 2,000–3,000 (yellow), 1,000–2,000 (blue), and <1,000 (teal). The second bar chart displays the logarithm of the total number of mapped reads for each tissue.
Analysis example: expression patterns of RT transcripts across tissues
The expression ratio between RT transcripts and their corresponding genes revealed pronounced tissue-specific differences in RT transcript expression (Fig. 2). It should be noted that genes lacking RT transcripts are not displayed in the figure. Therefore, the figure illustrates the expression relationship between genes with RT transcripts and their corresponding RT transcripts across different tissues, rather than depicting the expression patterns of all genes in these tissues. We have accordingly labeled the two distinct RNA-seq approaches (stranded vs. unstranded) to facilitate comparative analysis by researchers (Fig. 2).

Tissue-specific expression patterns of RT transcripts relative to their corresponding genes. The distribution of the expression ratio between RT transcripts and their corresponding genes, represented as log10(FPKM (RT transcript)/FPKM(Gene)), is shown for 43 tissues. The dashed vertical line at 0 indicates equal expression levels of RT transcripts and their parent genes. Negative values reflect the lower RT transcript expression relative to their genes, while positive values indicate higher RT transcript expression. The data from tissues marked with a star were derived from stranded RNA-seq libraries, while those without a marker came from unstranded RNA-seq libraries.
In most tissues, the distribution of expression ratios peaked below 0, indicating that RT transcripts are generally expressed at lower levels than their parent genes. However, the degree of this difference varied considerably across tissues. Notably, tissues such as the stomach, spleen, and small intestine (highlighted in red in Fig. 2) exhibited distributions closer to 0, suggesting that RT transcripts in these tissues are expressed at levels comparable to their corresponding genes. This observation may reflect the functional importance of RT transcripts in these transcriptionally dynamic tissues, where they might play roles in chromatin remodeling, the generation of alternative RNA isoforms, or other regulatory processes.
Conversely, tissues such as the testis, lung, and liver (highlighted in grey in Fig. 2) exhibited expression distributions with peaks significantly below −2, indicating that RT transcripts are expressed at markedly lower levels relative to their parent genes. This pattern suggests stringent regulation of TRT in these tissues, likely minimizing its impact on downstream genes and chromatin structure. The breadth of these distributions also varied across tissues. For example, the liver and thyroid displayed broader distributions, reflecting heterogeneity in RT transcript expression, with some transcripts achieving relatively high expression levels while others remained low. In contrast, tissues such as the kidney and pancreas exhibited narrower distributions, indicating more consistent and uniform RT transcript expression relative to their parent genes. These findings highlight substantial variability in RT transcript expression patterns across tissues, underscore the diverse roles of RT transcripts in gene regulation and their contribution to maintaining tissue-specific transcriptional equilibrium and provides a framework for deeper investigation into its biological significance.
Methods
Data collection
A comprehensive set of publicly available RNA-seq datasets and TRT data representing healthy human tissues was collected from National Center for Biotechnology Information (NCBI) and relevant published literature (https://doi.org/10.6084/m9.figshare.24848265.v3)22,25 (Supplementary Table S1). The human reference genome (GRCh38.p13) and corresponding gene annotation files (version 37) were obtained from the GENCODE database26.
Data analysis
The raw sequencing data underwent a rigorous quality control process to ensure reliability. Initially, the quality of raw RNA-seq reads was assessed using FastQC v0.12.1 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and the quality report was compiled with MultiQC v1.927. Low-quality bases and adapter sequences were removed using Trimmomatic v0.3928, employing default parameters alongside additional trimming thresholds for precision.
Subsequently, the high-quality reads were aligned to the reference genome using STAR v2.7.9a29. After alignment, TRT events were identified through ARTDeco30 with default parameters.
For the publicly available TRT data, all were derived from GTEx samples (https://doi.org/10.6084/m9.figshare.24848265.v3). Since GTEx samples were profiled using non-stranded RNAseq libraries, we filtered the results to report only entries that did not overlap with genes on the opposite strand, using the intersect function from bedtools (v2.30.0)31.
Additionally, we excluded the RT transcripts of non-expressed genes in each specific tissue. Expressed genes are defined as those with FPKM > 1 in at least 25% of the samples within a specific tissue.
For all identified TRT events, we used subseq function from seqtk (1.4-r122) (https://github.com/lh3/seqtk) to extract their sequences based on their positional information and the specific version of the genome downloaded above. The gene annotation information was extracted based on the Ensembl Gene ID from the following URL: https://grch37.ensembl.org/index.html and selected the median expression level across all samples as the data for plotting on the online platform.
Data Records
We have publicly shared the dataset on Figshare32 (https://doi.org/10.6084/m9.figshare.28974116) in CSV format, containing the following information for each Downstream-of-Gene (DoG) transcript:
Genomic localization details of both the gene (chromosome, start_position, end_position, strand) and the DoG transcript (chromosome (DoG), dog_start_position, dog_end_position, strand (DoG)).
Gene annotations (gene_id, Symbol, Synonym, Description) including functional descriptions and identifiers.
Sequence information (sequence) of the DoG transcript.
Tissues information (tissue) of the DoG transcript.
Average expression levels (mean-geneFPKM for the gene, mean-dogFPKM for the DoG transcript) across samples.
All expression data with per-sample values (all-geneFPKM, all-dogFPKM) and corresponding sample_ids.
Unique identifiers (DOG for the DoG transcript, gene_id for the associated gene).
Technical Validation
To systematically identify TRT events, we implemented a robust analytical pipeline using STAR-aligned BAM files and ARTDeco, a computational framework specifically designed for transcriptional readthrough characterization. ARTDeco employs a sliding window algorithm to detect continuous RNA-seq read coverage extending beyond the 3′ end of gene annotations by at least a default or user-defined minimum threshold. Transcriptional readthrough candidates were defined as regions meeting coverage thresholds across consecutive windows. To ensure analytical rigor, all downstream analyses exclusively utilized uniquely mapped reads identified through HOMER’s tools (v4.11)33, eliminating ambiguities from multi-mapped reads.
To maximize the reliability of TRT predictions, we implemented stringent quality control standards throughout all stages of data processing. To effectively mitigate batch effects, RNA-seq data were rigorously curated based on both tissue type and sequencing project criteria: for samples of the same tissue type, only those within the same study project were included, thereby completely avoiding interference caused by cross-project data integration. Recognizing that bidirectional transcriptional noise could confound TRT detection, we restricted analyses to strand-specific RNA-seq libraries. This critical filtering step enabled unambiguous assignment of transcriptional directionality, excluding signals from antisense transcription or overlapping genes on the reverse strand. Raw RNA-seq reads were first evaluated with FastQC, followed by comprehensive quality report generation using MultiQC. Subsequently, Trimmomatic was applied to filter out low-quality bases and adapter sequences, utilizing both default parameters and customized trimming thresholds to enhance processing accuracy.
For public TRT datasets derived from non-stranded GTEx libraries with ARTDeco, we implemented an additional validation layer using BEDTools (v2.30.0). Putative readthrough regions intersecting genes on the reverse strand were systematically excluded via bedtools intersect. Alignment and TRT detection tools between our pipeline and published TRT data were rigorously harmonized (STAR alignment, identical genome build, ARTDeco for prediction TRT). This methodological congruence enabled direct comparison while maintaining internal validity.
This multi-tiered quality control framework—spanning experimental design constraints, computational filtering, and directional specificity validation-ensured high-confidence TRT identification while addressing inherent limitations of transcriptional readthrough analyses in complex eukaryotic genomes.
Usage Notes
All data is directly downloadable from Figshare. Additionally, we have integrated these resources into our custom-developed online platform (http://www.hhrtbase.com/) that incorporates various analytical tools, providing convenient access for browsing, downloading, and utilization.
Code availability
No custom code was used. Software tools used for processing are mentioned in the Methods and Technical Validation sections.
References
Proudfoot, N. J. Transcriptional termination in mammals: stopping the RNA polymerase II juggernaut. Science 352, aad9926, https://doi.org/10.1126/science.aad9926 (2016).
Vilborg, A., Passarelli, M. C., Yario, T. A., Tycowski, K. T. & Steitz, J. A. Widespread inducible transcription downstream of human genes. Mol. Cell 59, 449–461, https://doi.org/10.1016/j.molcel.2015.06.016 (2015).
Hennig, T. et al. HSV-1-induced disruption of transcription termination resembles a cellular stress response but selectively increases chromatin accessibility downstream of genes. PLOS Pathog. 14, e1006954, https://doi.org/10.1371/journal.ppat.1006954 (2018).
He, H. et al. Long noncoding RNA ZFPM2-AS1 acts as a miRNA sponge and promotes cell invasion through regulation of miR-139/GDF10 in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 39, 159, https://doi.org/10.1186/s13046-020-01664-1 (2020).
Rutkowski, A. J. et al. Widespread disruption of host transcription termination in HSV-1 infection. Nat. Commun. 6, 7126, https://doi.org/10.1038/ncomms8126 (2015).
Alpert, T., Straube, K., Oesterreich, F. C., Herzel, L. & Neugebauer, K. M. Widespread transcriptional readthrough caused by Nab2 depletion leads to chimeric transcripts with retained introns. Cell Rep. 33, https://doi.org/10.1016/j.celrep.2020.108324 (2020).
Pessa, J. C., Joutsen, J. & Sistonen, L. Transcriptional reprogramming at the intersection of the heat shock response and proteostasis. Mol. Cell 84, 80–93, https://doi.org/10.1016/j.molcel.2023.11.024 (2024).
Hockel, M. & Vaupel, P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. JNCI J. Natl. Cancer Inst. 93, 266–276, https://doi.org/10.1093/jnci/93.4.266 (2001).
Wiesel, Y., Sabath, N. & Shalgi, R. DoGFinder: a software for the discovery and quantification of readthrough transcripts from RNA-seq. BMC Genomics 19, 597, https://doi.org/10.1186/s12864-018-4983-4 (2018).
Liang, D. et al. The output of protein-coding genes shifts to circular RNAs when the pre-mRNA processing machinery is limiting. Mol. Cell 68, 940–954.e3, https://doi.org/10.1016/j.molcel.2017.10.034 (2017).
Almarza, D. et al. Risk assessment in skin gene therapy: viral–cellular fusion transcripts generated by proviral transcriptional read-through in keratinocytes transduced with self-inactivating lentiviral vectors. Gene Ther. 18, 674–681, https://doi.org/10.1038/gt.2011.12 (2011).
Abe, K. et al. Downstream-of-gene (DoG) transcripts contribute to an imbalance in the cancer cell transcriptome. Sci. Adv. 10, eadh9613, https://doi.org/10.1126/sciadv.adh9613 (2024).
Choi, E.-S., Lee, H., Lee, C.-H. & Goh, S.-H. Overexpression of KLHL23 protein from read-through transcription of PHOSPHO2-KLHL23 in gastric cancer increases cell proliferation. FEBS Open Bio 6, 1155–1164, https://doi.org/10.1002/2211-5463.12136 (2016).
Pflueger, D. et al. Functional characterization of BC039389-GATM and KLK4-KRSP1 chimeric read-through transcripts which are up-regulated in renal cell cancer. BMC Genomics 16, 247, https://doi.org/10.1186/s12864-015-1446-z (2015).
Barresi, V. et al. Fusion transcripts of adjacent genes: new insights into the world of human complex transcripts in cancer. Int. J. Mol. Sci. 20, 5252, https://doi.org/10.3390/ijms20215252 (2019).
Vilborg, A. et al. Comparative analysis reveals genomic features of stress-induced transcriptional readthrough. The Proceedings of the National Academy of Sciences 114, E8362–E8371, https://doi.org/10.1073/pnas.1711120114 (2017).
Vo, T. V. et al. CPF recruitment to non-canonical transcription termination sites triggers heterochromatin assembly and gene silencing. Cell Rep. 28, 267–281.e5, https://doi.org/10.1016/j.celrep.2019.05.107 (2019).
Mylonas, C. & Tessarz, P. Transcriptional repression by FACT is linked to regulation of chromatin accessibility at the promoter of ES cells. Life Science Alliance 1, 1–14, https://doi.org/10.26508/lsa.201800085 (2018).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008, https://doi.org/10.1093/gigascience/giab008 (2021).
Li, Z., Qin, F. & Li, H. Chimeric RNAs and their implications in cancer. Curr. Opin. Genet. Dev. 48, 36–43, https://doi.org/10.1016/j.gde.2017.10.002 (2018).
Wu, H., Singh, S., Xie, Z., Li, X. & Li, H. Landscape characterization of chimeric RNAs in colorectal cancer. Cancer Lett. 489, 56–65, https://doi.org/10.1016/j.canlet.2020.05.037 (2020).
Caldas, P. et al. Transcription readthrough is prevalent in healthy human tissues and associated with inherent genomic features. Commun. Biol. 7, 1–12, https://doi.org/10.1038/s42003-024-05779-5 (2024).
Kim, P. & Zhou, X. FusionGDB: fusion gene annotation DataBase. Nucleic Acids Res. 47, D994–D1004, https://doi.org/10.1093/nar/gky1067 (2019).
Balamurali, D. et al. ChiTaRS 5.0: the comprehensive database of chimeric transcripts matched with druggable fusions and 3D chromatin maps. Nucleic Acids Res. 48, D825–D834, https://doi.org/10.1093/nar/gkz1025 (2020).
Sayers, E. W. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 50, D20–D26, https://doi.org/10.1093/nar/gkab1112 (2022).
Mudge, J. M. et al. GENCODE 2025: reference gene annotation for human and mouse. Nucleic Acids Res. 53, D966–D975, https://doi.org/10.1093/nar/gkae1078 (2025).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048, https://doi.org/10.1093/bioinformatics/btw354 (2016).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120, https://doi.org/10.1093/bioinformatics/btu170 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, https://doi.org/10.1093/bioinformatics/bts635 (2013).
Roth, S. J., Heinz, S. & Benner, C. ARTDeco: automatic readthrough transcription detection. BMC Bioinf. 21, 214, https://doi.org/10.1186/s12859-020-03551-0 (2020).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, https://doi.org/10.1093/bioinformatics/btq033 (2010).
Chen, X. Dataset for transcription readthrough events in healthy human tissues. figshare https://doi.org/10.6084/m9.figshare.28974116.v3 (2025).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589, https://doi.org/10.1016/j.molcel.2010.05.004 (2010).
Acknowledgements
We would like to express our sincere gratitude to the organizations and researchers who provided access to the public genomic data sets used in this study. And this work is supported by Zhejiang Provincial Natural Science Foundation (LQZQN25H250003).
Author information
Authors and Affiliations
Contributions
Y.M.: Writing–original draft, Resources, Formal analysis, Methodology, Visualization. Z.C. & Y.Q.: Formal analysis, Data curation, Resources. S.W.: Data curation, Resources. X.C.: Writing–review and editing, Methodology, Formal analysis, Data curation, Visualization, Project administration.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mei, Y., Cheng, Z., Lu, Y. et al. Comprehensive resource for transcription readthrough events in healthy human tissues. Sci Data 12, 1176 (2025). https://doi.org/10.1038/s41597-025-05557-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05557-w
