
Abstract
Compared to commercial sheep breeds, Tibetan sheep are exceptionally productive in high-altitude environments. Whole-genome sequencing can elucidate the genetic markers and candidate genes related to such economic traits, greatly accelerating the breeding progress. However, a lack of genome sequence data has hindered the functional gene dissection and genetic improvement of Tibetan sheep. This study presents whole-genome sequencing data from 301 Gamba sheep, a Tibetan national breed raised in extremely high-altitude regions. The data represents 12.3 Tb paired-end sequences generated on a BGI-T7 platform. An average sequencing depth of 13.8X, an average mapping ratio of 94.0%, and an average genome coverage of 99.6% illustrated the high quality of the sequence data. By aligning the sequence data to the sheep reference genome ARS-UI_Ramb_v3.0 with variants filtration, 39,718,985 single nucleotide polymorphisms (SNPs) and 5,275,473 insertions-deletions (InDels) were obtained. Genetic kinship analysis indicated the sample independence and high quality of the selected individuals. This whole-genome dataset significantly enriches public resources for studying the genetic diversity, genomic selection, and environmental adaptations of sheep.
Similar content being viewed by others

Background & Summary
The Tibetan Plateau is the highest altitude plateau on Earth, also known as the ‘Third Pole’1. Its southernmost and highest region is the Himalayan mountain chain, which has an average width of 200–350 km and a length of 2400 km. Here, the extremely cold and dry local climatic conditions combined with low oxygen levels have given rise to plants2 and animals3, including sheep, with excellent environmental adaptability, forming a unique ecosystem. Tibetan sheep broadly refers to all indigenous sheep breeds from the Tibetan Plateau region, which are mainly divided into the highland-, valley-, and oula-types4,5. As a breed of highland Tibetan sheep, the Gamba sheep have been shaped by long-term natural and artificial selection in the northern Himalayas6 and was recognized as a national-level breed in 2022. The breed is renowned for its uniquely flavoured mutton7 serving as a source of food and livelihood for locals. Despite its excellent adaptation to the harsh high-altitude environment, the breed’s low production efficiency still restricts economic development8,9. Unfortunately, due to the lack of genetic systematic characterization, the functional gene dissection and genetic improvement of Gamba sheep have been significantly hindered.
Despite this, research on Tibetan sheep breeds has expanded, focusing on two main topics of research namely genetic evolution and environmental adaptability. For example, Li et al. investigated their high-altitude adaptation10, while Sha et al. focused on ruminal microbiota-host gene interaction11. Based on multi-omics analyses, Han et al. found that the BMPR1B gene affects litter size in Tibetan sheep12, while Sun et al. investigated the genetic diversity and population structure of 11 Tibetan sheep populations13. The existing resequencing data of Tibetan sheep in public databases mainly stem from the hinterland of the Tibetan Plateau. As the national breed raised in the highest altitude county in China, Gamba sheep hold significant value in livestock genetic evolution and environmental adaptability studies. However, the breed’s whole genome sequence data still needs to be generated.
Next-generation sequencing technology enables high-throughput whole-genome sequencing (WGS) and it has wide applications in domestic animals, including tracing a species’ evolutionary trajectory or animal domestication14,15,16,17, assessing species-level biodiversity18, and identifying genes associated with animal economic traits19,20,21. Genomic analyses of samples from diverse ecological zones can elucidate the genetic underpinnings of local environmental adaptation, where beneficial genotypes can be identified can applied in improved productive breeds for superior performance under local environment.
Here we present new WGS data from 301 indigenous Tibetan sheep, encompassing 12.3 Tb of raw sequence data. These samples were sourced from the Himalayan region, specifically from five townships in Gamba County, Tibet Autonomous Region. Given the extreme environmental conditions of the sampling sites, with grazing areas situated at altitudes exceeding 5000 meters, this dataset constitutes the largest and highest-altitude WGS data generated from Tibetan sheep. The average sequencing depth of 13.8X in this dataset ensured the statistical power for genomic analyses and the sequencing data were aligned to the Ovis aries reference genome ARS-UI_Ramb_v3.022 (https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_016772045.2/). After performing variant calling and variant filtration, the Variant Call Format (VCF) files that comprise a total of 39,718,985 single nucleotide polymorphisms (SNPs) and 5,275,473 insertions/deletions (InDels) were obtained.
The data generated from this study are expected to aid in (1) Assessing genetic diversity to evaluate breed variability (e.g., calculating metrics such as nucleotide diversity and observed/expected heterozygosity); (2) Identifying signatures of positive selection to study genomic selection (e.g., comparing Gamba sheep with other Tibetan sheep); (3) Exploring genome-environment associations to study environmental adaptation (e.g., comparing high-altitude and low-altitude sheep breeds); (4) Exploring genome-trait associations for phenotypic and production traits (e.g., conducting genome-wide association studies); (5) Checking genetic variants from regions of interest (e.g., examining candidate genes or QTLs associated with production traits); (6) Constructing the Pan-genome of Tibetan sheep breeds; (7) Developing SNP genotyping chips for use in breeding programs; and (8) Studying the domestication histories of sheep in conjunction with sequencing data from other countries/regions. Overall, the large-scale whole-genome sequencing of Tibetan sheep from the Himalayan region provides a valuable resource for understanding the genetics of Tibetan sheep and enriching global sheep genome sequence databases.
Methods
Sample collection
This experiment was approved by the Animal Care and Use Committee of Southwest University (No. IACUC-20240920-09). A total of 301 Gamba sheep aged 1–5 years old (no breeding records, determined by experimenter based on body size and tooth wear) were selected from five towns (Gamba Town, n = 20; Longzhong Town, n = 100; Changlong Town, n = 52; Zhike Town, n = 35; Kongma Town, n = 94) of Gamba country, Tibet, China (Fig. 1a,b, Supplementary Table S1). All sheep were grazed naturally and supplemented with dry hay in barns. From each sheep, 3–5 mL jugular venous blood was sampled by a team of trained personnel and blood samples were anticoagulated with EDTA and stored at −20 °C for subsequent processing.

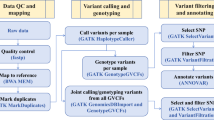
Tibetan sheep WGS sampling locations and study workflow. (a) The five sampling sites were situated in the Himalayan mountainous region, specifically on the border between China’s Gamba and India’s Sikkim. (b) The Gamba sheep flock, from which samples were collected, was housed in pens at an altitude of 4700 meters, however, during grazing, the flock can reach elevations as high as 4800–5000 meters. (c) Workflow: Blood samples were collected from 301 Gamba sheep and subjected to whole-genome sequencing, generating a total of 12.3 Tb of sequencing data. The genome processing pipeline encompassed quality control, reads mapping, variant calling, variant filtering, and variant annotation.
Whole genome sequencing
The 301 blood samples were prepared for DNA extraction using the genomic DNA extraction kit (QT-1001, IGENEBOOK, China). Before library construction, the concentration, integrity, and purity of each genomic DNA sample were assessed using 1.5% agarose gel electrophoresis and a NanoDrop spectrophotometer (Thermo Scientific, USA). Qualified genomic DNA samples were randomly broken into fragments of about 150 bp, end-repaired, and ligated to adapters, followed by purification with magnetic beads and PCR amplification using the KAPA HiFi HotStart DNA Polymerase (Kapa Biosystems, USA). Then, the libraries were denatured to single-stranded DNA, circularized, digested to linear DNA, and quantified using a Qubit Fluorometer (Thermo Scientific, USA). The qualified DNA library was sequenced by the IGENEBOOK company (Wuhan, China) using the DNBSEQ-T7 platform.
Genomic alignment and variant calling
This study employed a rigorous genomic analysis pipeline, which mainly includes quality control, reads mapping, variant calling, variant filtering, and variant annotation (Fig. 1c). The original off-machine data in fastq files were quality-controlled using the fastp tool (v0.23.423), and sequence alignment and variant detection were performed using the Sentieon Genomics software (v20230824,25). In brief, the clean reads were aligned to the sheep reference genome sequence (ARS-UI_Ramb_v3.022, https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_016772045.2/) using the bwa software (v0.7.1726). The BAM files were sorted, and duplicates were marked using the Picard package (v2.25, https://broadinstitute.github.io/picard) and the mapping rate and genome coverage were calculated using Samtools (v1.1327) and PanDepth (v2.2528), respectively. The Sentieon haplotyper module was used to call variants for each sample to independently generate a genomic Variant Call Format (gVCF) file, while the joint variant calling was carried out by the Sentieon GVCFtyper module from the 301 gVCF files, to finally create a common VCF file. The SNP and InDel variates were extracted and hard filtrated using the SelectVariants module in GATK (v4.1.8.129), and were then further filtered using VCFtools (v0.1.1630).
Variant annotation and genetic kinship analysis
The filtered SNPs and InDels were functionally annotated using the snpEff software (v.5.131), and the variant locations of the intronic, untranslated, upstream, downstream, and intergenic regions were calculated. The variant counts and cumulative proportions were calculated to observe variant depth, thereby reflecting variant quality. The kinship matrix was constructed based on whole-genomic SNPs using the GEMMA software (v0.98.532) and visualized using the R package heatmap (v1.0.12). The kinship result was used to observe whether samples were independent of each other, thereby reflecting the sample quality.
Data Records
The original 301 sheep whole genome sequencing data in FASTQ format have been deposited in the Genome Sequence Archive33 on the China National Center for Bioinformation (CNCB) platform, under accession number CRA02448334 (https://ngdc.cncb.ac.cn/gsa/browse/CRA024483). The variation data generated including the final SNP.vcf and InDel.vcf files were deposited in the Genome Variation Map35 on the CNCB platform, under accession number GVM00101336 (https://ngdc.cncb.ac.cn/gvm/getProjectDetail?project=GVM001013).
Technical Validation
Quality control for sequencing data
Based on the high-throughput sequencing of 301 Gamba sheep, we obtained 12,289.3 Gb of raw data, where for each sample, 188 to 593 million reads and 28.2 Gb to 88.9 Gb sequencing yield (number of bases generated) were obtained. As shown in Table 1 and Fig. 2a, the sequencing depth for the samples varied from 9.5X to 30.5X (averaging 13.8X), with a GC content average of 43.0%, of which 96.0–99.3% and 88.2–96.8% of the bases had a minimum Phred scaled quality score of 20 (Q20, sequencing error rate < 0.01) and 30 (Q30, sequencing error rate < 0.001), indicating a high expected base calling accuracy. The quality reports (Fig. 2b,c) confirmed the overall high-quality scores of all the sequencing reads. As shown in Fig. 2d,e, the genome coverage and properly mapped rates of the sequence reads against the sheep reference genome (ARS-UI_Ramb_v3.0) was 99.6% (from 99.4% to 99.7%) and 94.0% (from 78.2% to 98.8%), respectively. These indicators confirmed the high quality of the sequencing data from multiple aspects.

Statistics of the high-throughput sequencing data of 301 Tibetan sheep and their alignment to the sheep reference genome. (a) Boxplots showing the sequencing yield, reads, depth, phred quality scores (Q20 and Q30), and GC content of 301 indigenous Tibetan sheep. (b) Mean quality value across each base position in a 150 bp read. (c) The sequence quality score plot shows that the quality scores of almost all reads fell between 35 and 40. (d) The genomic coverage of sequencing reads on the 26 autosomes of the sheep reference genome. (e) The mapping rate of sequencing reads to the sheep reference genome. Each circle or line represents one sample of the 301 Tibetan sheep dataset.
Quality control of SNP and InDel data
Joint genotyping of all samples originally identified 48,691,893 SNPs and 7,449,956 InDels. To ensure variant quality and minimize false positives, variant filtration was performed using the GATK software29. A series of statistical metrics, including Mapping Quality (MQ), Quality by Depth (QD), Fisher Strand (FS), Strand Odds Ratio (SOR), Mapping Quality Rank Sum Test (MQRankSum), and Read Position Rank Sum Test (ReadPosRankSum) were used to evaluate variant quality. These metrics assess aspects such as coverage depth, and alignment quality at variant positions, and detect strand bias detection collectively filtering potential false positives and ensuring accurate variant calling. The SNPs and InDels were filtered out when the missing genotype rate of a variant exceeded 10% in all samples and, a final 39,718,985 SNPs and 5,275,473 InDels were retained (Fig. 3a,b).

Statistics of variants depth and annotation in WGS data from 301 Tibetan sheep. After variant annotation, the proportions of SNPs (a) and InDels (b) at specific chromosomal location categories were statisticed. The cumulative distribution statistics demonstrated the high quality of the SNP (c) and InDel (d) data. Each line represents one sample of the 301 Tibetan sheep dataset.
Summary statistics of SNPs and InDels
High-quality variants were distributed across the genome with an average density of 1 SNP every 67 bases and 1 InDel every 456 bases (Table 2). Although the number of SNPs was approximately seven-fold higher than that of InDels, their variant classes were similar. As shown in Fig. 3a,b, most SNPs and InDels were located in intronic regions, accounting for about 70% of the total, while only 1.7% of SNPs and 1.2% of InDels were located in Exons, and approximately 2% of variants were located in the UTR regions. The arithmetic average of variant counts for all SNPs and InDels were 11.8 and 11.6 respectively (Fig. 3c,d), while the cumulative depth distribution plots illustrated the high quality of the identified SNPs and InDels.
Genetic kinship of all samples
Based on whole genomic SNPs, the relatedness coefficients for any two animals were calculated. Figure 4a shows the heatmap of the kinship matrix, and no animals were directly related to one another, with 90% and 95% of the relatedness coefficients lower than 0.011 and 0.022 (Fig. 4b). The results of the genetic kinship analysis demonstrated the independence of the samples and the high quality of the selected samples in the whole-genome resequencing dataset.

Genetic kinship analysis of 301 Tibetan sheep WGS data indicated satisfactory sample independence. (a) Genetic relationship matrix of 301 sheep based on genome-wide SNPs; (b) The histogram frequency distribution of the pairwise kinship coefficients.
Usage Notes
The large-scale whole-genome sequencing dataset was derived from Gamba sheep, a national breed from the Himalayan region. Tibetan sheep is a general term that refers broadly to all indigenous sheep breeds from the Tibetan Plateau and as one of the Tibetan sheep breeds, Gamba sheep significantly enrich the global genomic resources of sheep. The study released both raw sequencing data and processed variant files. Notably, the choice of reference genome can to some extent affect the variant quality and downstream analysis37,38. The variants we presented were based on the reference genome of the Rambouillet breed (ARS-UI_Ramb_v3.022), although a Tibetan sheep reference genome, CAU_O.aries_1.039 (https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_017524585.1/) was assembled by Prof. Li’s team previously. It may be more appropriate to align data to the CAU_O.aries_1.0 reference when conducting studies only focused on Tibetan sheep. This year, Prof. Li’s team also published the T2T-sheep1.040 (https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_040805955.1/), the first Telomere-to-Telomere genome of the renowned Hu sheep breed. Compared to traditional reference genomes, the T2T reference genome fills gaps in repetitive sequences, particularly in complex regions such as centromeres and subtelomeres. These genome versions provide greater flexibility for addressing diverse research objectives related to Tibetan sheep in future studies.
Code availability
Genome analyses were conducted by standard bioinformatic tools running on a Linux operating system. To dramatically accelerate the genome analysis process, the Sentieon Genomics software24 (https://www.sentieon.com/) that is faithful to GATK Best Practices29 (http://www.broadinstitute.org/gatk/guide/best-practices) was employed. Please note that Sentieon is a commercial software that requires a license, which can be obtained by purchasing or applying for a one-month free trial. The main steps, software, and code/parameters used from off-machine data to the VCF file are described below.
(1) Quality control of raw reads
Software: fastp v0.23.4
Code: fastp_install_dir/fastp -w 8 -i sample.R1.fq -I sample.R2.fq -o sample.R1.clean.fq -O sample.R2.clean.fq
(2) Mapping reads and sorting the BAM file
Software: Sentieon v202308
Code: Sentieon_install_dir/bin/sentieon bwa mem -R “@RG\tID:rg_sample\tSM:sample\tPL:$PL” -t 40 -K 10000000 sheep.genome. fa sample.R1.clean.fq sample.R2.clean.fq | util sort -t 40–sam2bam -o sample.sorted.bam -i -
(3) Mark Duplicates
Software: Sentieon v202308
Code: Sentieon_install_dir/bin/sentieon driver -t 40 -i sample-sorted.bam --algo LocusCollector --fun score_info sample.score.txt
Sentieon_install_dir/bin/sentieon driver -t 40 -i sample-sorted.bam --algo Dedup --score_info sample.score.txt--metrics sample.dedup_metrics.txt sample.deduped.bam
(4) Sequencing accuracy assessment and alignment statistics (optional)
Software: Sentieon v202308, PanDepth v2.25, and Samtools v1.13
Code for calculating reads quality:
Sentieon_install_dir/bin/sentieon driver -r sheep.genome.fa -t 40 -i sample-sorted.bam \
--algo MeanQualityByCycle sample.mq_metrics.txt \
--algo QualDistribution sample.qd_metrics.txt \
--algo GCBias--summary sample.gc_summary.txt sample.gc_metrics.txt \
--algo AlignmentStat sample.aln_metrics.txt \
--algo BaseDistributionByCycle sample.bd_metrics.txt \
–algo QualityYield sample.qy_metrics.txt \
--algo InsertSizeMetricAlgo sample.is_metrics.txt)
Code for calculating genome coverage:
PanDepth_install_dir/pandepth -i sample.deduped.bam -t 8 -o genome.coverage.stat.txt
Code for calculating mapping rate:
Samtools_install_dir/Samtools flagstat -@40 -i sample.deduped.bam > sample.mapped.stat.txt
(5) Variants calling for each sample
Software: Sentieon v202308
Code: Sentieon_install_dir/bin/sentieon driver -r sheep.genome.fa -t 40 -i sample.deduped.bam–algo Haplotyper--emit_conf = 30 --call_conf = 30 --emit_mode gvcf sample.gvcf.vcf.gz
(6) Variant joint calling for all samples
Software: Sentieon v202308
Code: GVCF_list = gvcf.gz.list.txt #paths to all gVCF files
GVCF_inputs = $(awk ‘{info = info” -v “$0} END {print info}’ $GVCF_list)
Sentieon_install_dir/bin/sentieon driver -t 40 -r sheep.genome.fa–algo GVCFtyper $GVCF_inputs joint-calling.vcf
(7) Variants extraction and hard filtration
Software: GATK v4.1.8.1
Code for SNPs:
GATK_install_dir/gatk SelectVariants -R sheep.fa -V joint-calling.vcf -O filtered.SNP.vcf \
-select ‘((QD>=2.0 && MQ>=40.0 && FS<=60.0 && SOR<=3.0) && (QD>=2.0 & & MQ<40.0 && FS<=60.0 && SOR<=3.0 || MQRankSum>=−12.5 && ReadPosRankSum>=−8.0) && vc.isSNP())’
Code for InDels:
GATK_install_dir/gatk SelectVariants -R sheep.fa -V joint-calling.vcf -O filtered.InDel.vcf \
-select ‘((QD>=2.0 && FS<=200.0 && SOR<=10.0) && (QD > = 2.0 & & FS<=00.0 && SOR<= 10.0 || ReadPosRankSum>=−20.0) && vc.isIndel())’
(8) Further filtration
Software: VCFtools v0.1.16
Code for SNPs:
Vcftools_install_dir/vcftools --vcf filtered.SNP.vcf --max-alleles 2 --min-alleles 2 --min-meanDP 5 --max-missing 0.9 --remove-filtered-all --recode --recode-INFO-all --out SNP.vcf
Code for InDels:
Vcftools_install_dir/vcftools --vcf filtered.InDel.vcf --min-meanDP 5 --max-missing 0.9–remove-filtered-all --recode --recode-INFO-all --out InDel.vcf.
References
Yao, T. et al. Third Pole Environment (TPE). Environmental Development 3, 52–64, https://doi.org/10.1016/j.envdev.2012.04.002 (2012).
Bala, E. et al. Five Himalayan weeds as potential bioresources for bioactive agents: toxic compounds to valuable scaffolds. Phytochem. Rev. 23, 1205–1246, https://doi.org/10.1007/s11101-024-09921-3 (2024).
Jiao, X. et al. Landscape Heterogeneity Explains the Genetic Differentiation of a Forest Bird across the Sino-Himalayan Mountains. Mol. Biol. Evol. 41, https://doi.org/10.1093/molbev/msae027 (2024).
Hu, L. et al. Genome-wide analysis of CNVs in three populations of Tibetan sheep using whole-genome resequencing. Frontiers in Genetics 13, https://doi.org/10.3389/fgene.2022.971464 (2022).
Song, Y. et al. Genome-Wide Selection Signals Reveal Candidate Genes Associated with Plateau Adaptation in Tibetan Sheep. Animals 14, https://doi.org/10.3390/ani14223212 (2024).
Ding, H. et al. Effects of different rearing systems on carcase traits, physicochemical properties, basic chemical composition, fatty acid profiles and amino acid profiles of Gangba lamb. Italian Journal of Animal Science 23, 362–372, https://doi.org/10.1080/1828051x.2024.2314156 (2024).
Zhang, Q. et al. Gangba sheep in the Tibetan plateau: Validating their unique meat quality and grazing factor analysis. Journal of Environmental Sciences 101, 117–122, https://doi.org/10.1016/j.jes.2020.06.024 (2021).
Jize, Z. et al. Different feeding strategies can affect growth performance and rumen functions in Gangba sheep as revealed by integrated transcriptome and microbiome analyses. Frontiers in Microbiology 13, https://doi.org/10.3389/fmicb.2022.908326 (2022).
Liu, X. et al. Dynamic changes in the gastrointestinal microbial communities of Gangba sheep and analysis of their functions in plant biomass degradation at high altitude. Microbiome 13, https://doi.org/10.1186/s40168-024-02022-5 (2025).
Li, X. et al. Whole-genome resequencing to investigate the genetic diversity and mechanisms of plateau adaptation in Tibetan sheep. Journal of Animal Science and Biotechnology 15, https://doi.org/10.1186/s40104-024-01125-1 (2024).
Sha, Y. et al. Response of Ruminal Microbiota-Host Gene Interaction to High-Altitude Environments in Tibetan Sheep. International Journal of Molecular Sciences 23, https://doi.org/10.3390/ijms232012430 (2022).
Han, B. Y. et al. Multiomics Analyses Provide New Insight into Genetic Variation of Reproductive Adaptability in Tibetan Sheep. Mol. Biol. Evol. 41, https://doi.org/10.1093/molbev/msae058 (2024).
Sun, L. et al. Resequencing reveals population structure and genetic diversity in Tibetan sheep. BMC Genomics 25, https://doi.org/10.1186/s12864-024-10800-6 (2024).
Zhao, Y.-X. et al. Genomic Reconstruction of the History of Native Sheep Reveals the Peopling Patterns of Nomads and the Expansion of Early Pastoralism in East Asia. Mol. Biol. Evol. 34, 2380–2395, https://doi.org/10.1093/molbev/msx181 (2017).
Yang, J. et al. Structural variant landscapes reveal convergent signatures of evolution in sheep and goats. Genome Biology 25, https://doi.org/10.1186/s13059-024-03288-6 (2024).
Fang, W. et al. Novel insights into sheep domestication in Southwest Asia based on genome-wide sequencing. Anim. Genet. 56, https://doi.org/10.1111/age.13500 (2025).
Cheng, H. et al. Long divergent haplotypes introgressed from wild sheep are associated with distinct morphological and adaptive characteristics in domestic sheep. PLoS Genet. 19, https://doi.org/10.1371/journal.pgen.1010615 (2023).
An, Z. X., Shi, L. G., Hou, G. Y., Zhou, H. L. & Xun, W. J. Genetic diversity and selection signatures in Hainan black goats revealed by whole-genome sequencing data. Animal 18, https://doi.org/10.1016/j.animal.2024.101147 (2024).
Zhang, J., Xiao, M., Fang, J., Huang, D. & Zhao, Y. Phenotypic, transcriptomic, and genomic analyses reveal the spatiotemporal patterns and associated genes of coarse hair density in goats. Zool. Res. 46, 825–840, https://doi.org/10.24272/j.issn.2095-8137.2025.034 (2025).
Zhang, J., Fang, J., Zhang, S., Xu, J. & Zhao, Y. Several variants on chromosome 10 are associated with coarse hair diameter in Dazu black goats (Capra hircus). Anim. Genet. 56, https://doi.org/10.1111/age.13509 (2025).
Li, R. et al. A sheep pangenome reveals the spectrum of structural variations and their effects on tail phenotypes. Genome Res. 33, 463–477, https://doi.org/10.1101/gr.277372.122 (2023).
Bickhart, D. M. et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 49, 643–650, https://doi.org/10.1038/ng.3802 (2017).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Freed, D., Aldana, R., Weber, J. A. & Edwards, J. S. The Sentieon Genomics Tools - a fast and accurate solution to variant calling from next-generation sequence data. bioRxiv 115717, https://doi.org/10.1101/115717 (2017).
Aldana, R. & Freed, D. Data Processing and Germline Variant Calling with the Sentieon Pipeline. Methods in molecular biology (Clifton, N.J.) 2493, 1–19, https://doi.org/10.1007/978-1-0716-2293-3_1 (2022).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Yu, H., Shi, C., He, W., Li, F. & Ouyang, B. PanDepth, an ultrafast and efficient genomic tool for coverage calculation. Briefings in Bioinformatics 25, https://doi.org/10.1093/bib/bbae197 (2024).
McKenna, A. et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303, https://doi.org/10.1101/gr.107524.110 (2010).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158, https://doi.org/10.1093/bioinformatics/btr330 (2011).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92, https://doi.org/10.4161/fly.19695 (2012).
Zhou, X. & Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 44, 821–824, https://doi.org/10.1038/ng.2310 (2012).
Chen, T. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genomics Proteomics & Bioinformatics 19, 578–583, https://doi.org/10.1016/j.gpb.2021.08.001 (2021).
CNCB Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA024483 (2025).
Li, C. et al. Genome Variation Map: a worldwide collection of genome variations across multiple species. Nucleic Acids Res. 49, D1186–D1191, https://doi.org/10.1093/nar/gkaa1005 (2021).
CNCB Genome Variation Map https://ngdc.cncb.ac.cn/gvm/getProjectDetail?project=GVM001013 (2025).
Rick, J. A., Brock, C. D., Lewanski, A. L., Golcher-Benavides, J. & Wagner, C. E. Reference Genome Choice and Filtering Thresholds Jointly Influence Phylogenomic Analyses. Syst. Biol. 73, 76–101, https://doi.org/10.1093/sysbio/syad065 (2024).
Li, H. et al. Exome variant discrepancies due to reference-genome differences. Am. J. Hum. Genet. 108, 1239–1250, https://doi.org/10.1016/j.ajhg.2021.05.011 (2021).
Li, X. et al. Genomic analyses of wild argali, domestic sheep, and their hybrids provide insights into chromosome evolution, phenotypic variation, and germplasm innovation. Genome Res. 32, 1669–1684, https://doi.org/10.1101/gr.276769.122 (2022).
Luo, L.-Y. et al. Telomere-to-telomere sheep genome assembly identifies variants associated with wool fineness. Nat. Genet. 57, https://doi.org/10.1038/s41588-024-02037-6 (2025).
Acknowledgements
This work was supported by the Gamba County black sheep population (group) breeding project.
Author information
Authors and Affiliations
Contributions
Jipan Zhang: Conceptualization, Sample collection, Data Curation, Formal Analysis, Visualization, Writing-Original Draft; Yangsheng Zhang: Sample collection; Mengying Wang: Sample collection; Luobu Gesang: Sample collection; Duoji Zhaxi: Sample collection; Ciren Pubu: Sample collection; Jikang Zheng: Sample collection; Hong Miao: Sample collection, Funding Acquisition; Yongju Zhao: Sample collection, Supervision, Funding Acquisition. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, J., Zhang, Y., Wang, M. et al. Whole-genome sequencing resources of 301 indigenous Tibetan sheep from the Himalayan region. Sci Data 12, 1351 (2025). https://doi.org/10.1038/s41597-025-05650-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05650-0
