
Abstract
The hawthorn spider mite, Amphitetranychus viennensis, is a major pest of orchards and ornamentals in the Palaearctic region, with adaptability and acaricide resistance. The lack of high-quality genomic resources limits understanding of its detoxification mechanisms and the development of RNAi-based pest control strategies. In this study, we utilized Illumina, Pacific Biosciences (PacBio), and Hi-C sequencing technologies to assemble a chromosome-level reference genome of A. viennensis. The assembled genome spans 141.96 Mb, with a contig N50 of 1.35 Mb. BUSCO analysis confirmed a high level of completeness, covering 91.6% of annotated genes. The assembly includes 50.97 Mb of repetitive sequences, representing 35.93% of the genome, and annotates 13,968 protein-coding genes. Using Hi-C sequencing, we anchored 47 contigs to three chromosomes, accounting for 97.27% of the estimated nuclear genome and achieving a contig N50 of 45.83 Mb. This high-quality genome assembly provides a valuable foundation for evolutionary and genomic research on spider mites, while also serving as a genetic resource to inform molecular control strategies and support sustainable pest management.
Similar content being viewed by others


Background & Summary
The hawthorn spider mite, Amphitetranychus viennensis (Arachnida, Acari, Acariformes, Trombidiformes, Tetranychidae), one of the most destructive pests affecting various host plants within Rosaceae family, distributes mainly in Europe and Asia1,2,3. A. viennensis feeding on plant juice from leaves and young buds causes yellow spots, leaf curling, defoliation, and ultimately a decrease in the photosynthetic capacity of plants2. Synthetic chemicals have been extensively utilized for controlling A. viennensis, consequently leading to the development of resistance in A. viennensis to nearly all commercially available acaricides4,5,6,7. The development of pesticide resistance, combined with pesticide residues in both food products and the environment6,8, has led to search for alternative pest management strategies, including the emerging biotechnology of RNAi9,10,11,12,13,14. Genome information is helpful for managing pesticide resistance and developing control strategies to tackle agricultural pests. Lack of genomic resources in A. viennensis becomes a limiting factor for such efforts. To enhance the management of pesticide resistance, as well as to facilitate the incorporation of RNAi into the existing integrated pest management strategies, we have sequenced the genome and transcriptomes of A. viennensis.
In this paper, we aim to assemble a high-quality chromosome-level genome of A. viennensis through combined application of Illumina, PacBio sequencing, and Hi-C data. The genome assembly consisted of 243 contigs with a total length of 141.96 Mb, of which the contig N50 was 1.31 Mb. In addition, 97.27% of the draft assembly was anchored to 3 chromosomes with a scaffold N50 of 45.83 Mb. A total of 13968 protein-coding genes were obtained, of which 94.16% were annotated. We also identified 16.70 Mb of DNA repeats, accounting for 11.78% of the genome assembly. The high-quality chromosome-level genome assembly of A. viennensis will provide a genetic basis for further research on this pest mite.
Methods and Results
Sample collection
The hawthorn spider mite was collected from crabapple Malus ‘Radiant’ tree in Tai’an City, Shandong Province, China (117.1194°E, 36.1964°N), and was subsequently reared with fresh peach, Prunus davidiana, in climate incubators at a temperature of 26 ± 0.5°C with a relative humidity of 50% and a photoperiod of 16 L: 8 D. Ten generations were reared and the eggs of the tenth generation were collected following the procedures in Tetranychus urticae15 for sequencing.
Library construction and genome sequencing
Genomic DNA was extracted from eggs using CTAB methods for Illumina, PacBio and Hi-C sequencing to prevent contamination from other individuals and microorganisms. Total RNA was extracted from a mixed sample containing mites across all developmental stages (eggs, larvae, adult females, and adult males) using the TRIzol reagent, followed by transcriptome library preparation and sequencing. The purity and integrity of genomic DNA and RNA were validated by the NanoDrop 2000C spectrophotometer (Thermo, Wilmington, DE, USA), and further assessed through fragment analyzer and 1.5% agarose gel electrophoresis, respectively.
For next-generation sequencing, the high-quality genomic DNA was fragmented into target fragments (350 bp) using ultrasonic shock. Subsequently, these fragments were utilized to construct short-read sequencing libraries. After removing adapter sequences and low-quality reads, a total of 32.47 million clean reads (Table 1) were obtained for subsequent analyses. For PacBio sequencing, PCR-free Single-Molecule Real Time (SMRT) library constructed following the manufacturer’s standard instructions subsequently was sequenced on a PacBio Sequel II platform (Berry Genomics Company, Beijing, China)16. Through quality control, 1,834,436 reads were obtained in total, with an average read length of 10.083 kb (Table 1). The Hi-C library constructed through standard instructions were performed on the Illumina Novaseq 6000 platform by Berry Genomics Company (Berry Genomics Company, Beijing, China), and 69.29 million reads of 150-bp paired-end clean reads were obtained (Table 1). RNA-seq libraries for transcriptome sequencing were constructed by Biomarker Technologies (Beijing, China) and then sequenced on the Illumina HiSeq 2000 platform, yielding 10.32 Gb of 150-bp paired-end reads.
Genome survey and assembly
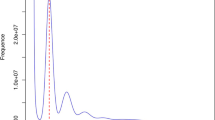
The main genome characteristics, including genome size, repetitive sequence content and heterozygosity, were essential to estimate before assembly. The k-mer (K = 19) frequencies were constructed based on Illumina clean short-reads using Jellyfish v1.1.1117. The estimated genome scale of A. viennensis was 157.65 Mb, with a heterozygosity of 0.317% (Fig. 1A). Subsequently, the draft genome was assembled using PacBio SMRT raw reads by Falcon software (length_cutoff = 8000, length_cutoff_pr = 12000)18,19. To further improve the quality and accuracy of the genome assembly, we corrected the genome by long-read and short-read polishing with Arrow (SMRT link version v5.0.1) and Pilon v1.16 with default parameters20,21. Then redundant heterozygous contigs were removed via Redundans v0.14a with default parameters22. As a result, we generated a 141.96 Mb genome assembly consisted of 243 contigs with the contig N50 of 1.31 Mb. For chromosome-level assembly, 69,288,927 Hi-C clean reads were obtained after filtering out sequences containing ≥3 unidentified nucleotides (Ns), adapter contamination, or ≥20% low-quality bases (Phred score ≤ 5), using default parameters. To verify the absence of exogenous contamination, 10,000 randomly selected clean reads were aligned against the NCBI non-redundant nucleotide database (NT, 2023 release) using BLASTN v2.12.0 (E-value cutoff: 1e−5). The full set of Hi-C clean reads was then aligned to the draft genome assembly using Juicer v1.6.223,24. Chromosomal scaffolding was performed with 3D-DNA v180922 using default settings, and manual corrections were made by inspecting Hi-C contact heatmaps in Juicebox v1.9.8 to resolve potential misassembles25,26. Finally, 47 scaffolds were anchored to 3 chromosomes (Fig. 2A,B) with a scaffold N50 of 45.83 Mb, covering a span of 141.96 Mb and representing 97.27% of the draft genome assembly.

Genome scope profiles of 19-mer analysis and GC content and depth distribution of A. viennensis genome. (A) Genome scope profiles of 19-mer analysis; (B) GC content and depth distribution of A. viennensis genome.

Genome assembly of A. viennensis. (A) Heatmap of genome-wide Hi-C data. The heatmap shows all interactions between 3 chromosomes, the frequency of interaction links is represented by the color, which ranges from yellow (low) to red (high), the redder, the higher of the intensity; (B) Overview of the genomic landscape of A. viennensis. Blocks on the outmost circle represent all 3 chromosomes, peak plots from outer to inner circles in green, red, and purple represent gene distribution, distribution of repeating elements, GC content and inter-chromosomal collinearity, respectively.
Assessment of assembly completeness was generated using BUSCO v3.0.2b27. The results showed that 91.6% of BUSCO genes could be successfully detected, of which 82.1% are single-copied and 9.5% are duplicated (Table 2).
Genomic repeat annotation
The prediction of repeat elements has been carried out using a combination of de novo and homology-based methods28,29,30. The detailed workflow was as follows: (1) MITE-Hunter v1.0 was used to perform de novo prediction of miniature inverted-repeat transposable elements (MITEs) in the A. viennensis genome assembly to construct a MITE library31. (2) LTRharvest and LTR Finder v1.07 were used to de novo detect long terminal repeat (LTR) sequences, and LTR_retriever v2.9.0 was employed to integrate the results and build an LTR library32,33,34. (3) RepeatMasker v4.1.1 was used to identify conserved repetitive elements by performing a homology-based search against the RepBase database (release 20181026)35,36. (4) The known repeats identified in step (3) were merged with the MITE and LTR libraries to create a custom repeat library, which was then used with RepeatMasker v4.1.1 to mask repetitive regions in the genome. (5) Finally, RepeatModeler v2.0.2a was employed to de novo identify additional repetitive sequences in the genome after the initial masking37. Ultimately, we identified 47.81 Mb interspersed repeats and 3.16 Mb tandem repeats. Among classified interspersed repeats, DNA transposons were the most abundant with a whole length of 16.70 Mb (Table 3).
Gene prediction and functional annotation
Three approaches, including de novo prediction, homolog-based and transcriptome-based methods were combined to perform gene prediction after eliminating the interference of repeat sequences in A. viennensis genome. The de novo gene models were predicted using software tools of AUGUSTUS v3.5.0, SNAP (http://snap.stanford.edu/snap/download.html), Glimmerhmm v3.0.4 and GeneMark-ET v4.32 in the repeat-masked genome38,39,40,41. Homology-based gene prediction was conducted using GeMoMa v1.9 against the protein sequences of Tetranychus urticae (GCF_000239435.1), Ixodes scapularis(GCF_016920785.2), Galendromus occidentalis (GCF_000255335.2), and Sarcoptes scabiei (GCA_020844145.1) downloaded from GenBank and then exon and intron boundary information was obtained through the comparison between the transcript and the genome42. For transcriptome-based gene prediction, HISAT v2.2.1 was used to align RNA-Seq reads to the genome sequence and Cufflinks v2.2.1 was used to assemble transcripts for obtaining the full-length transcript sequences43,44. PASA software v2.5.2 was used to predict the open reading frame based on the obtained full-length transcript sequence45. EvidenceModler (https://sourceforge.net/projects/evidencemodeler/) was used to integrate the above prediction results, and untranslated region (UTR) and other variable cut annotation was predicted by PASA v2.5.246. As a result, 13968 protein-coding genes with a mean coding sequence length of 1613 bp were identified from A. viennensis genome (Table 4). All protein-coding genes were aligned to three integrated protein sequence databases: NR, SwissProt, eggNOG. Protein domains were annotated by InterproScan v5.66–98.0 and the Gene Ontology (GO) terms for each gene were obtained from the corresponding eggnog-mapper annotation entry47. The pathways in which the genes might be involved were assigned by BLAST v2.12.048 against the KEGG databases. The protein-coding gene functional annotation results were merged from the above methods. Finally, 94.16% (13152/13968) of protein-coding genes were annotated (Table 5). Furthermore, 8617 genes were assigned with GO terms and 8158 genes were mapped to at least one KEGG pathway.
For the annotation of non-coding RNA tRNA was annotated by tRNAscan-SE v2.0.11. The Rfam database was used to annotate other types of ncRNA by BLAST v2.12.0 software.
Data Records
The Illumina, PacBio and Hi-C sequencing data that were used for the genome assembly and annotation have been deposited in the NCBI Sequence Read Archive with accession number SRP50018649. The final chromosome assembly has been deposited at GenBank under the accession number GCA_050437165.150. Genome annotation file and RNA-seq data are available at the Figshare database51.
Technical Validation
We comprehensively evaluate the quality of genome assembly by evaluating the quality of the sequencing data, the completeness of the assembly, and the correctness of the assembly: 1) Upon aligning clean reads from genomic sequencing to the genome assembly with BWA, an impressive data response ratio of 98.99% was achieved. This high alignment rate indicates robust mapping of the majority of sequencing reads to the reference genome, highlighting the efficacy of the analysis; 2) no scatter clusters appear in the depth distribution map of GC content (Fig. 1B), indicating that there is no contamination in the splicing results; 3) the BUSCO evaluation showed that 91.6% of BUSCO genes (single-copy gene: 82.1%, duplicated gene: 9.5%) were successfully identified in genome assembly, indicating that the genome assembly was very complete; 4) the Hi-C heatmap revealed a well-organized interaction contact pattern along the diagonals within/around the chromosome inversion region, which indirectly confirmed the accuracy of the chromosome assembly.
Code availability
No custom code has been used for the research. Data processing was performed by the relevant pipelines and software in accordance with the manual, and protocols and versions, as well as useful parameters, have been described in the methods section. The default parameters suggested by the developers were used in those pipelines and software unless specifically indicated.
References
Kafil, M., Allahyari, H. & Saboori, A. Effect of host plants on developmental time and life table parameters of Amphitetranychus viennensis (Acari: Tetranychidae). Experimental and Applied Acarology. 42, 273–281 (2007).
Yang, J. et al. Target gene selection for RNAi-based biopesticides against the hawthorn spider mite, Amphitetranychus viennensis (Acari: Tetranychidae). Pest Management Science. 79, 2482–2492 (2023).
Kasap, İ. Life history of hawthorn spider mite Amphitetranychus viennensis (Acarina Tetranychidae) on various apple cultivars and at different temperatures. Experimental and Applied Acarology. 31, 79–91 (2003).
Li, D. X., Tian, J. & Shen, Z. R. Assessment of sublethal effects of clofentezine on life-table parameters in hawthorn spider mite (Tetranychus viennensis). Experimental and Applied Acarology. 38, 255–73 (2006).
Grbić, M. et al. The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature. 479, 487–492 (2011).
Van Leeuwen, T. & Dermauw, W. The molecular evolution of xenobiotic metabolism and resistance in chelicerate mites. Annual Review of Entomology. 61, 475–498 (2016).
Wang, X., Zhao, X. & Tu, H. Study on the variations in acaricide sensitivity between two spider mite species, Amphitetranychus viennensis and Tetranychus urticae, in Chinese orchards. Pesticide Biochemistry and Physiology. 210, 106367 (2025).
De Rouck, S., Inak, E., Dermauw, W. & Van Leeuwen, T. A review of the molecular mechanisms of acaricide resistance in mites and ticks. Insect Biochemistry and Molecular Biology. 159, 103981 (2023).
Baum, J. A. et al. Control of coleopteran insect pests through RNA interference. Nature Biotechnology. 25, 1322–1326 (2007).
Schutter, K. D. et al. RNAi-based biocontrol products: market status, regulatory aspects, and risk assessment. Frontiers in Insect Science. 1, 818037 (2022).
Zhang, J., Khan, S. A., Khan, S. A. & Bock, R. Next-generation insect-resistant plants: RNAi-mediated crop protection. Trends in Biotechnology. 35, 871–882 (2017).
Mao, Y. B. et al. Silencing a cotton bollworm P450 monooxygenase gene by plant-mediated RNAi impairs larval tolerance of gossypol. Nature Biotechnology. 25, 1307–1313 (2007).
Head, G. P. et al. Evaluation of SmartStax and SmartStax PRO maize against western corn rootworm and northern corn rootworm: efficacy and resistance management. Pest Management Science. 73, 1883–1899 (2017).
Rodrigues, T. B. et al. First sprayable double-stranded RNA-based biopesticide product targets Proteasome subunit beta type-5 in Colorado potato beetle (Leptinotarsa decemlineata). Frontiers in Plant Science. 12, 728652 (2021).
Suzuki, T. et al. Protocols for the delivery of small molecules to the two-spotted spider mite, Tetranychus urticae. PLoS One. 12, e0180658 (2017).
Chin, C.-S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature Methods. 10, 563–569 (2013).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764–770 (2011).
Kingan, S. B. et al. A high-quality de novo genome assembly from a single mosquito using PacBio sequencing. Genes (Basel). 10, 62 (2019).
Chaisson, M. J. & Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics. 13, 238 (2012).
Krajaejun, T. et al. PacBio long read-assembled draft genome of Pythium insidiosum strain Pi-S isolated from a Thai patient with pythiosis. BMC Research Notes. 16, 271 (2023).
Hu, J. et al. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 36, 2253–2255 (2020).
Sato, R. et al. The first released available genome of the common ice plant (Mesembryanthemum crystallinum L.) extended the research region on salt tolerance, C (3)-CAM photosynthetic conversion, and halophilism. F1000Research. 12, 448 (2023).
Durand, N. C. et al. Juicer Provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell systems. 3, 95–98 (2016).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Genomics, 1–3 (2013).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell systems. 3, 99–101 (2016).
Simão, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212 (2015).
Jiang, S. D. et al. Chromosome-level genome assembly of a stored-product psocid, Liposcelis tricolor (Psocodea: Liposcelididae). Scientific Data. 11, 1310 (2024).
Liu, Y. et al. A chromosome-level genome assembly of tomato pinworm, Tuta absoluta. Scientific Data. 10, 390 (2023).
Xu, C. et al. Chromosome level genome assembly of oriental armyworm Mythimna separata. Scientific Data. 10, 597 (2023).
Han, Y. & Wessler, S. R. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Research. 38, e199 (2010).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics. 9, 18 (2008).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research. W265-8 (2007).
Ou, S. & Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiology. 176, 1410–1422 (2018).
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics. 4.10.1–4.10.14 (2004).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics. 25, 4–10 (2009).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences of the United States of America. 117, 9451–9457 (2020).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research. 34, (Web Server issue), W435-9 (2006).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics. 5, 59 (2004).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 20, 2878–2879 (2004).
Lomsadze, A., Burns, P. D. & Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Research. 42, e119 (2014).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods in Molecular Biology. 1962, 161–177 (2019).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology. 37, 907–915 (2019).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 28, 511–515 (2010).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research. 31, 5654–5666 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biology. 9, R7 (2008).
Cantalapiedra, C. P. et al. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Molecular Biology and Evolution. 38, 5825–5829 (2021).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of Molecular Biology. 215, 403–410 (1990).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP500186 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_050437165.1 (2025).
Ren, M. et al. Chromosome-level genome assembly of hawthorn spider mite, Amphitetranychus viennensis (Acari: Tetranychidae). figshare. Dataset. https://doi.org/10.6084/m9.figshare.28052918 (2025).
Acknowledgements
This project was supported by the earmarked fund for Modern Agro-industry Technology Research System (No.2025CYJSTX18), Shanxi scholarships for Science and Technology Activities for Overseas students (No.20230019), Excellent PhD Scientific Research Startup Project (No. 2023BQ123), and the National Natural Science Foundation of China (No.32102224).
Author information
Authors and Affiliations
Contributions
X.Z. and J.Y. designed the project. M.R., Y.Z., Y.W. and Y.D. conducted the sampling and sequencing. M.R., Y.Z. and Y.W. performed the comparative genomics analysis, gene family identification. M.R. and J.Y. drafted the manuscript, and Y.G., Z.L., P.Z., L.H. and R.F. improved and revised the manuscript. All authors have read and approved its final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ren, M., Zhang, Y., Wang, Y. et al. Chromosome-level genome assembly of hawthorn spider mite, Amphitetranychus viennensis (Acari: Tetranychidae). Sci Data 12, 1342 (2025). https://doi.org/10.1038/s41597-025-05683-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05683-5
This article is cited by
-
Genome size estimation and evolution trends in Acari
Experimental and Applied Acarology (2025)
