
Abstract
Species extinctions in the tropics are accelerating, outpacing documentation efforts. Meanwhile, DNA barcoding is flourishing in the Global North, backed by extensive infrastructure, allowing non-taxonomic experts to identify species from nonlethal, minimally invasive, and environmental samples. However, hyper-diverse regions like Peru make up only 0.52% (n = 93,246) of the Barcode of Life Database (BOLD). To address this, we established three decentralized laboratories with low-cost, portable nanopore sequencers. From 2018–2023, we generated 1,858 barcodes in situ using six genetic markers for 1,097 vertebrates and 76 plants from existing and new biobanks. We present the first genetic barcodes for 30 mammal and 196 bird species from Peruvian specimens, increasing the number of Peruvian mammal and bird species in BOLD by 110% and 36.5% respectively. We also report the first records of the marsupial Marmosops ocellatus and the bat Sturnira lilium for Peru. This dataset represents an effort to go from fresh or museum-preserved samples to barcodes entirely in situ, avoiding the export of samples outside the country, and facilitating local capacity in molecular biodiversity research.
Similar content being viewed by others

Background & Summary
Species richness alone does not fully capture biodiversity patterns, especially when assigning priority regions for conservation1. However, understanding species richness can lead to significant advances in ecological, biogeographical, and evolutionary research2. It constitutes a crucial step in the multicriteria decision process for prioritizing conservation efforts, including the designation of protected areas and the allocation of conservation funding3. This is because species richness is more easily understood by the public and policymakers. Currently, biodiversity counts face several significant challenges. First, a slow species identification process is hindered by the declining number of scientists capable of identifying wild organisms4,5 and limited access to training and financial support for specializations in systematics and taxonomy, known as the “taxonomic impediment”6. Second, species populations are rapidly declining and extinction rates are accelerating due to systemic threats. For instance, 80% of major global armed conflicts between 1950–2000 occurred within biodiversity hotspots7. The Living Planet Index, which monitors over 16,000 wildlife populations, estimates a 60% global decline in wildlife population sizes and an 89% decline in Central and South American species over a 44-year period since 19708. Third, there is a geographic bias towards industrialized countries in temperate regions9,10 leaving potentially millions of taxa in high-diversity biomes facing extinction before they are fully described11,12,13,14,15. Advanced technologies like DNA barcoding16, portable low-cost sequencing equipment17,18, and in situ training on these technologies19,20 can help to reduce these shortages in local expertise and scientific resources.
Over the last two decades, advancements in DNA sequencing technology have accelerated the acquisition of sequences for potential “universal” DNA barcodes, or genetic markers, as tools for rapid species identification21,22. This approach, known as DNA barcoding, has expanded the number of researchers who can participate in species identification using nonlethal, noninvasive or minimally invasive, and environmental samples23,24,25,26,27,28. However, the effectiveness of DNA barcoding for species identification relies on a large, well-curated, and open-access DNA reference database16,29,30,31. In response to this need, the Consortium for the Barcode of Life (CBOL) was established in 200416 bringing together over 200 organizations from 50 countries to publish DNA barcodes in a single, publicly accessible curated database.
Today, the Barcode of Life Data System (BOLD) serves as the primary repository for the acquisition, storage, validation, and analysis of DNA barcodes, advancing our understanding of planetary biodiversity16,32. However, genetic biomonitoring is heavily biased by geography and taxonomy, creating large regional taxonomic skews and complicating the identification process. For example, in the Americas, as of July 15, 2024, the USA and Mexico have generated and deposited three times as many DNA records in BOLD (525,792 sequences for ~ 40,000 species) compared to similarly megadiverse South American countries (Brazil, Colombia, Ecuador, Peru, and Venezuela), with only 165,721 sequences for around 8,000 species combined. Given that South American species richness is high33, it is clear that the availability of local research and sequencing infrastructure greatly influences the likelihood of generating genetic records for a country’s biodiversity. This disparity often forces countries to export samples to foreign institutions for costly sequencing services, which can range from $7 USD per gene (e.g. at Macrogen today) or nearly $10.80 USD per species identified in an eDNA sample in 202127. In addition, all exports incur additional bureaucratic hurdles that increase the time and cost for species identification, and limit opportunity for in-country scientists to conduct sequencing locally and operate on international timescales for data generation34,35.
DNA barcoding has traditionally relied on first-generation sequencing techniques, primarily Sanger sequencing developed in 197736,37. Second-generation sequencers have been tentatively adopted by some barcoding efforts38,39, but these are not cost-effective without highly multiplexed sampling, nor are they equitably distributed given that second-generation sequencing technologies are both physically large and out of reach of many budgets inside biodiversity hotspots. Recently, third-generation sequencing has leveled the playing field. Oxford Nanopore Technology’s MinION MK1b, MK1c, and MK1d which are the size of a modern smartphone, along with advances in portable miniaturized equipment for PCR (quantitative or standard), mini centrifuges, self-imaging electrophoresis systems, lyophilized reagents, and minimal cold chain requirements, have enabled DNA sequencing to be conducted with basic infrastructure17,40,41,42,43. Nanopore sequencing has been efficiently deployed in challenging field conditions to address time-sensitive biodiversity questions17,43,44,45,46. The learning curve for this technology is not steep, making it useful in classrooms for hands-on learning in molecular biology19,47. Additionally, it has proven to be both efficient and cost-effective in high-throughput applications22,48. Most projects have utilized either in situ Sanger sequencing or high-throughput sequencing, but rarely an in situ high-throughput sequencing approach.
Here, we present results from a collaborative, high-throughput, in situ, and museum-based sequencing effort in Peru. Peru is one of the 10 megadiverse countries in the world, with 573 mammals, 1,894 birds, 637 reptiles, 674 amphibians, 1,607 fishes, and 20,825 known plant species49,50,51,52,53,54. Despite this diversity, Peru is genetically under-surveyed and underrepresented. For example, when you consider genetic sequences derived from Peruvian specimens, only 55% (180 of 326) of Amazonian mammal species have at least one genetic marker, while that number drops to only 4.3% (65 of 1,506) for bird species55,56. Moreover, Peru accounts for only 0.5% of total records in BOLD, which spans approximately 1,000 vertebrate species and 8,000 species of plants and invertebrates. Our study increases the genetic representation of the Amazonian region of Peru, within the Western Amazonian Forests and Plains bioregion (NT18) (Oneearth.org), and standardizes a method across several Peruvian stakeholders for efficiently closing geographic and taxonomic species gaps. We utilized three biosample sources: 1) field surveys by biologists in the department of Madre de Dios, 2) wildlife rescue centers, and 3) tissue biobanks with curated specimens at the Natural History Museum of San Marcos University (Lima, Peru) from 12 Peruvian Amazonian departments. This effort marks the first high-throughput DNA barcoding of diverse taxa conducted entirely in-country using real-time portable sequencing technologies. We barcoded faunal specimens of birds, mammals, reptiles, and amphibians with universal DNA markers (COI, cytochrome b (cyt b), and 18S) and flora, which often require multiple markers for reliable identification (matK, rbcL, trnH-psbA)57,58. In this dataset, we present: 1) a curated dataset of DNA barcodes for a broad range of Peruvian Amazonian taxa in a Barcode of Life (BOLD) repository, 2) an in situ field laboratory sample-to-sequence DNA barcoding pipeline, 3) a cost-effective approach driven by a multiplexing strategy validated on multiple nanopore chemistries, and 4) a bioinformatics pipeline that minimizes online resource usage, maximizes sample identification reliability, is flexible across any genetic marker and counters common field laboratory contamination. We demonstrate the utility of in situ laboratory systems to efficiently and cost-effectively address geographic biases in DNA barcoding efforts and support the expansion of curated DNA reference repositories.
Methods
Study sites and sampling
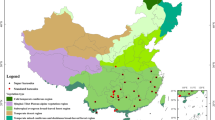
Biosamples for this study were obtained from three sources: field surveys, a museum biobank representing collections from various sites, and, to a lesser extent, samples donated from rescued wildlife at rehabilitation centers (Fig. 1). Overall, samples were collected from the lowland forests of the Loreto, Madre de Dios, Ucayali, and Puno departments of Peru (See File 1: collection sites available at Figshare59) for details on collection sites). These include 122 collection sites across the following Peruvian ecoregions: Bosque Húmedo Amazónico (32.23%), Bosque Muy Húmedo Montano (22.31%), Bosque Muy Húmedo Premontano (30.58%), Bosque Pluvial Montano (7.44%), Bosque Seco Oriental (3.31%), Bosque Seco Estacional (1.65%), Puna Húmeda Seca (1.65%), and Meso-Andino (0.83%)60.

A map of sample localities for vertebrates and plants collection in Peru and the three in situ laboratories that conducted the sequencing in this study. 1) Museo de Historia Natural of the Universidad Nacional Mayor de San Marcos, Lima (MHN-UNMSM); 2) the GreenLab at the Amazon Field Station by Inkaterra (GL), Madre de Dios; 3) the Los Amigos Wildlife Conservation Laboratory (WCL), Madre de Dios. Map produced in ArcGIS Pro v3.2.0, using basemaps from ESRI and Peruvian ecoregions60. Sampling localities are listed in File 1: collection sites available at Figshare59.
Field surveys
The Department of Madre de Dios (MDD) of southeastern Peru (85,300 km2, 200–4,000 m a.s.l.) harbors 62,040 km2 of federally protected forest, including the Manu, Bahuaja-Sonene, and Alto-Purus National Parks, and the Tambopata National Reserve61. The department is almost entirely covered by humid primary rainforest at 200–400 m above sea level and receives an average of 300 cm of precipitation per year, with daily temperatures ranging from 20–30 °C61. Surveys for biosamples took place within the lowland rainforests of MDD between May 2018 and June 2023 in two formats: 1) during a vertebrate mark-recapture program operated by Field Projects International at the Los Amigos Conservation Concession (LACC), and 2) during a plant survey within the Inkaterra Ecological Reserve. Birds, mammals, reptiles, and amphibians were biosampled, marked, and released as part of the One Health biosurveillance program of the In Situ Laboratory Initiative (https://insitulabs.org/) at the LACC. Samples included frozen blood stored in Longmire’s buffer62, blood in 1X DNA/RNA Shield Buffer (Zymo Research, R1200), feathers stored dry at room temperature, frozen skin biopsies in a 500 uL of lab-made RNA storage buffer63, and feathers stored dry with silica gel. Additionally, a targeted survey was conducted of 110 plant specimens representing 13 known families and 84 unique taxa at the Inkaterra Ecological Reserve. For plants, fresh samples of a 30–40 cm portion of a lateral branch with leaves were collected per plant. Detailed protocols for animal handling and tissue collection can be found at Protocols.io for work with birds64, bats65, non-volant mammals66,67,68, and reptiles and amphibians69.
Rescue center sampling
The Taricaya Eco Reserve (12°31′ S, 68°58′ W) and Amazon Shelter (12°38.9′ S, 69°12.5′ W) contributed nine blood samples and one biopsy sample from 10 individuals across four species (Alouatta sara, Ateles chamek, Lagothrix lagotricha, and Tamarinus imperator) between 2021–2023. Samples were obtained during annual health checks and included hair stored in Ziploc bags with desiccant, as well as blood in Longmire’s solution62 and on FTA elute cards (Cytiva, Marlborough, MA). The animals are suspected to have originated in the wild from areas in the immediate vicinity of the rescue centers. However, due to the nature of the rescues, the exact geographic location of each individual cannot be determined with certainty.
Sample sterility for field-derived specimens
Standard protocols to maintain sample sterility and prevent disease transmission from or to sampled fauna were followed by collectors. These included the use of face masks during handling, changing gloves between study subjects, using new sterile autoclaved collection tubes, and a four-step sterilization protocol for biopsy punches and tweezers between uses (immersion in 10% bleach, two distilled water rinses, 70% alcohol, and air drying on a sterile surface). Detailed safety measures are also available in our taxa-specific collection protocols64,65,66,67,68,69.
Museum samples
For museum specimens, we sampled from material currently deposited at the mammal and bird scientific collections in the Museum of Natural History of the National San Marcos University, MHN-UNMSM (Lima, Perú). We took biopsies from skin preparations, feathers, or used preserved muscle tissue stored in ethanol (97°) at −20 °C.
Ethical statement on sampling
In this dataset, we utilized biobanked tissues at the MHN-UNMSM, an entity affiliated with the Servicio Nacional Forestal y de Fauna Silvestre (SERFOR) as an official, registered biobank. Additional field surveys conducted for this study employed mark-recapture methodology to sample each organism in a minimally invasive way, taking detailed morphological and photographic evidence to ensure reliable field identifications. Wildlife handling procedures followed the Animal Behaviour Society Guidelines70 and the American Society of Mammalogists’ Guidelines on wild mammals in research71. SERFOR granted annual research and collection permits for these field surveys (N° 403-2016-SERFOR-DGGSPFFS, N° 193-2015-SERFOR-DGGSPFFS, N° 245-2018-SERFOR-DGGSPFFS, N° 171-2019-MINAGRI-SERFOR-DGGSPFFS, N° 888-2008-INRENA-ATFFS-TAMBOPATA-MANU). Additionally, the Animal Studies Committee of the University of Missouri–St. Louis and the San Diego Zoo Wildlife Alliance approved all protocols (1208181-3, 23-011).
The In Situ laboratory network
Sample storage and processing occurred at three laboratory sites. The GreenLab (GL, 2018-) is located 17 km east of Puerto Maldonado (PEM) on the Madre de Dios River at the Amazon Field Station by Inkaterra (Fig. 1). It was set up collaboratively by the Inkaterra Association and Field Projects International at a materials cost of less than $12,000 by sourcing used items and in-kind donations to limit new product purchases. The Los Amigos Wildlife Conservation Laboratory (WCL, 2021-) is situated at the Estación Biológica Los Amigos (EBLA), formerly known as el Centro de Investigación y Capacitación Río Los Amigos (CICRA), approximately 94 km west of PEM (Fig. 1). It was established collaboratively by the Amazon Conservation Association, Conservación Amazónica, San Diego Zoo Wildlife Alliance and Field Projects International, with foundational support from the Gordon and Betty Moore Foundation.
Both the GL and WCL are true rainforest field laboratories, accessible only by boat and powered by a combination of solar and diesel generators. While the GL is a single-room establishment suitable for DNA barcoding studies19, the WCL has three separate sterile spaces for high-throughput handling for disease surveillance at a biosafety level of 2. The third laboratory in the network is located in Lima at the MHN-UNMSM (2022-) within the Mammalogy and Avian scientific collections. The laboratory at MHN-UNMSM was set up by the San Diego Zoo Wildlife Alliance in conjunction with the Museum staff under the support of a El Consejo Nacional de Ciencia, Tecnología e Innovación (CONCYTEC) grant. Both the WCL and the MHN-UNMSM laboratories have high-throughput automated liquid handlers that can extract nucleic acids from 96 specimens in under six hours. All sample handling, processing, and sequencing was conducted by Peruvian, Latinamerican, and visiting research scientists at each laboratory.
The sample processing pipeline
We present an end-to-end solution for high-throughput sample processing, DNA barcoding, and sequencing using portable sequencing technology alone (Fig. 2). The method outlined in Fig. 2 is the one we advocate other groups use, and corresponds to a particular indexing strategy that we found to work the best. The complete pipeline is outlined below, many steps of which are supported by protocols published on Protocols.io by the WCL laboratory team and others40.

An infographic of the laboratory analysis pipeline using a dual indexing strategy (Method C).
DNA extraction
Samples were lysed according to tissue-specific protocols72,73. DNA was extracted from 300 µL of lysate per sample into two elutions (50 µL and 30 µL). The automated extraction protocols are documented on Protocols.io (protocols.io/insitulabs) and the slight variations by tissue type and laboratory are captured in Tables 1–3. All extracts were quantified on a Quantus fluorometer (Promega, Madison, WI) and stored at −20 °C for further use. All elutions generated by magnetic bead kits were cleaned up using SPRI beads at 0.8x ratio74.
Gene markers and primers
Three mitochondrial genetic markers were amplified for vertebrates (Table 4): cytochrome oxidase subunit I (COI), cytochrome b (cyt b), and 18S.
COI was amplified with a primer cocktail for mammals (C_VFILFt1, C_VRLRt1)72 and primer pair AmphF2_t1-AmphR3_t1 for herpetofauna75. Primers MTCB-F and MTCB-R76 or MVZ05 and MVZ1677 were used for the cyt b gene. For birds, primers BirdF178 and COIbirdR279 were used to amplify the COI gene, and primers L14996.gyps1 and H379.gyps80 for the cyt b gene. For all vertebrates tested, two primer pairs (G3 and G4) were used to amplify the 18S gene81.
Plants were screened for the following chloroplast barcodes: matK, with primers MatK-390f and MatK-1326r82, trnH-psbA with the primers trnH-283 and psbA3 f84, and rbcL, with the primers rbcLa-F85 and rbcLa-R86. All primers were modified before use by adding a universal ONT adaptor to the 5′ end of the forward (TTT CTG TTG GTG CTG ATA TTG C) and reverse (ACT TGC CTG TCG CTC TAT CTT C) primers to make them compatible with PCR-based ONT barcoding strategies. Primer details and adapter sequences for the ONT barcodes are in Table 5.
Amplification of genetic markers
At the GL,12.5 µL reactions were used of 0.1 µM of each forward and reverse primer, 1X buffer, 1.5 mM MgCl2, 0.05 mM each dNTP, and 0.3 units of GoTaq Hot Start DNA Taq Polymerase (Promega, Madison, WI). At the WCL and MHN-UNMSM labs, 12.5 or 15 µL final reactions were used, comprising 1X GoTaq G2 Hot Start Green Master Mix, 0.5X of each 10 µM primer, and 2 µL of DNA template. Specific PCR mixes and cycling conditions for each gene marker are provided in Table 6 and the automated protocol for PCRs is on Protocols.io87.
Indexing strategies
The pipeline for library preparation involves indexing in one of three ways in each lab - Method A at the GL, Method B for Batch 1 of sequecing and Method C for Batch 2 of sequencing at the WCL, and Method C at MHN-UNMSM. These methods correspond to the chronological evolution of our indexing strategy at these labs over time and reflect demultiplexing ease and an attempt to reduce costs. We now utilize Method C as the preferred indexing strategy for future projects.
Method A utilized the PCR barcoding expansion 1–96 kit (Oxford Nanopore Technologies EXP-PBC096), which contains 96 barcodes. To accommodate all samples, we multiplexed samples within each barcode group. Samples from distinct taxonomic orders were pooled, with an average of 4.01 ± 0.47 samples (range 3–5) and 2.21 ± 0.42 samples (range 2-3) per barcode in Libraries 1 and 2, respectively, to proportionately reflect the number of amplicons they contained. This strategy has been replaced by Method C.
Method B applied a combinatorial indexing approach with 16 ONT-fw primers and combined each of them with 19 ONT-rv primers making a total of 304 indexes. To multiplex even higher numbers, for each index pair, we included 2–4 amplicons that included COI and cyt b genes of different taxa (herpetofauna, bat or terrestrial mammal) that are distinguishable bioinformatically. The indexing primers were custom ordered to match those in the EXP-PBC096 kit. Indices were annealed using a 15 µL PCR volume, with 1 µL of each barcode, 0.2 mM dNTPs, 1X buffer, 2.5 mM MgCl2, and 0.075 units of GoTaq Hot Start DNA Taq Polymerase (Promega, Madison, WI). The indexing PCR involved an initial denaturation for 2 mins at 95 °C, followed by 18 cycles at an annealing temperature of 62 °C. Each indexed product was quantified (1 µL), normalized based on the number of samples they contained (drawing more PCR product from samples multiplexed to a greater extent), and pooled into a single tube in equimolar proportions. We generated a custom SPRI-bead cleanup solution using 1 mL of carboxylated Sera-Mag™ Magnetic SpeedBeads in a binding buffer of 10 mM Tris base, 1 mM EDTA, 2.5 M NaCl, 20% PEG 8000, and 0.05% Tween 20 at pH 8.088. The libraries were purified using this SPRI-bead cleanup in a 1:0.8x ratio. This strategy has been replaced by Method C.
Method C employed a dual-indexing strategy with custom-engineered 24 bp long indices matching EXP-PBC096. A second indexing PCR was conducted to attach each index to an amplicon at annealing temperature of 55 °C, and a maximum of 15 cycles, and then cleaned, as in Method B. Once we had pools of up to 96 samples, a Native Barcode from the Oxford Nanopore Technologies NBD114.24 kit was ligated to the outside of each pool. This ensured that every amplicon had a unique inner and outer index combination, and this protocol is published online89.
Sequencing
Sequencing was conducted in situ at each of the three labs on MK1b and MK1c nanopore sequencers over the duration of the project (Table 7). At GL and the MHN-UNMSM lab, some sequences were produced as part of training workshops with students and staff19. Library preparation for sequencing involved normalizing all pools proportional to the number of amplicons they contained, standardizing to ~200 fmols for a set of 96 samples. Once normalized, each library pool underwent end-prep and dA-tailing using the NEBNext® Ultra™ II End Repair/dA-Tailing Module (New England Biolabs, E7546) and adaptor ligation using the NEBNext Quick Ligation Module (New England Biolabs, E6056) as per ONT protocols. In Method C, we also included the ligation of Native Barcodes using the NEB Blunt/TA Ligase Master Mix (New England Biolabs, M0367) before adaptor ligation.
Throughout the project, ONT’s sequencing chemistries and flowcells have evolved significantly, requiring us to adapt our sequencing protocols. Two libraries at GL were sequenced on R9 flowcells with SQK-LSK108 chemistry, while a third used SQK-LSK109 chemistry. At WCL, all libraries were sequenced with SQK-LSK110 chemistry, and at MHN-UNMSM, all libraries were sequenced with SQK-LSK114, the most current sequencing chemistry. Overall, R9 flowcells were used at GL and WCL, and R10.4.1 flowcells were used at MHN-UNMSM.
Basecalling and demultiplexing sequence data
The full pipeline is publicly available90 and uses publicly available custom Docker images designed to eliminate the majority of software compatibility issues during installation on different operating systems90. The bioinformatics pipeline can be run on a device with a 1Tb SSD and 16 GB RAM, which are the minimum requirements to run a MK1b portable sequencer, over the course of no more than 3 days.
We used slightly different initial basecalling pipelines on the data, as both data formats produced by ONT and basecalling algorithms evolved over the course of the project. For the earliest sequencing data produced at the GL, multi-fast5 files were first converted back into single fast5 files using the ONT_fast5_api (https://github.com/nanoporetech/ont_fast5_api) to merge data from all runs into the same pipeline. For runs at the WCL, outputs were in multi-fast5 format, while runs at MHN-UNMSM produced output in pod5 formats.
The files generated by GL and WCL were basecalled using Guppy with a high accuracy model (v6.3.5), whereas the MHN-UNMSM runs were basecalled on Dorado 0.2.1 and 0.5.1 using a super accuracy model (SUP) (dna_r10.4.1_e8.2_400bps_fast@v4.1.0). Nanoplot91 was used to evaluate run quality and read lengths overall. Raw reads were filtered for quality (q score > 5) and read length (>150 bp) using Nanofilt (NanoFilt, RRID:SCR_016966) for all data. Filtered reads were then demultiplexed using Guppy Barcoder with primer trimming enabled and modifications to a dual barcoding configuration as needed to fit custom indices (GL and WCL, v6.1.2; MHN-UNMSM, v6.4.6).
Creating consensus sequences per barcode
To create consensus sequences from data produced by all runs at all three labs, we used the NGSpeciesID pipeline92 with a 30X minimum read coverage cutoff for numbers of reads required per consensus sequence90. Briefly, NGSpeciesID is a reference-free clustering and consensus pipeline for long-reads based on isONclust93 that forms a de novo reference and polishes it repeatedly to reduce errors. The pipeline has been validated for wildlife forensics applications94,95, with MinION consensus sequences displaying either no difference or very small differences in comparison to Sanger-generated control sequences. This study demonstrated that none of the tested MinION consensus sequence replicates deviated from their corresponding Sanger sequence by more than a single base pair per ~420 bps of sequence95. A detailed comparison of Nanopore-based DNA barcoding with Sanger sequencing can also be found here. Sequencing chemistries and raw-read accuracies have improved significantly since this study, which was conducted on R9 chemistry, and further validations are underway for these new chemistries (R10 and above) (Vasiljevic, personal communication). In short, MinION sequencing is now being adopted heavily for larger DNA barcoding efforts, including the sequencing of 100,000 amplicons on a single MinION run by the Centre for Biodiversity Genomics in Guelph, with over 95% of these sequences being identical to those produced on a PacBio Sequel II96. R10 chemistries have been further validated in metabarcoding pipelines, where single raw read accuracies matter much more than in DNA barcoding pipelines, and found to average at 99% accurate97.
Ascribing sequence IDs to consensus sequences
Validated consensus sequences (see Technical Validation) were cross-referenced against the nt GenBank (release 228) and BOLD databases using nucleotide blast (blast + 2.12.0; “blastn” with parameters ‘-remote -db nt -outfmt “7 std stitle” -max_target_seqs. 10 -evalue 1e-6’) and BOLD’s online identification engine (Fig. 3). Accession number and species identity of each top reference hit were merged with the data of each indexed sample (in indexing strategies A and B), matching sequences to their taxonomic groups based on BLAST output.

A flowchart of the consensus sequence screening performed to quality check each sequence before accepting it into the final dataset.
Once a final curated dataset was created, specimen data were uploaded to BOLD [DS-ISLPE24]98 and GenBank along with individual sequences for each amplicon. All metadata, including images curated per species (either live in hand during field surveys, or from specimens at the museum), were then attached to the completed dataset, and shared on Figshare59.
Data Records
The dataset is available at BOLD [DS-ISLPE24]98. This reference dataset contains the following information: 1) sample ID, 2) catalog number (for Museum specimens), 3) collection date, 4) field ID, 5) voucher type, 6) collection site coordinates when available, 7) names of specialists who provided the field ID, and 8) images from multiple angles when available. For each genetic marker, the following information is also available: 1) gene name, 2) primers utilized, including their sequences, 3) sequence length, and of course, 4) the genetic sequence of the barcode itself. All barcode sequences, specimen metadata, primer metadata, and specimen images from this study are stored on BOLD. The sequences have also been published on GenBank under Accession numbers: PV251947-PV251993 for matk, PV251903-PV251946 for rbcL, PV391297-PV391873 for cyt b, PV402776-PV403636 for COI, PQ887451-PQ887718 for 18S, and PQ884100-PQ884132 for trnH-psbA. Since these sequences are generated by high-throughput sequencing and not Sanger sequencing, there is no trace file (the spectrogram produced with typical Sanger sequencing). BOLD relies heavily on this to assign if a sequence is barcode compliant, a standard that cannot be met with high-throughput sequencing techniques of any platform, including the nanopore sequencing we utilized here. Thus, barcode compliance cannot be achieved for this or any modern dataset generated by multiplexing samples on a high-throughput sequencer; however, we are hopeful that future iterations of BOLD will be able to provide modified requirements that make barcode compliance a possibility.
We successfully generated 1830 barcodes for 430 species from 1169 individuals across 35 orders, 81 families, and 303 genera (Fig. 4). Of these, 239 specimens were barcoded at the GL, 577 specimens at the WCL, and 357 specimens at the MHN-UNMSM. The majority of the specimens derived from the MDD, although 11 other departments were also represented in this dataset (Fig. 4). This dataset includes 50 specimens of amphibians, 497 birds, 534 mammals, and 16 reptiles. Within the dataset, 835 specimens had at least one barcode that produced a genetic ID that matched the field identification, and this was used to declare the species name for the specimen. The distribution of barcodes generated across each marker for the classes are listed in Table 8.

A representation of the percent of individuals analysed (a) per Department of Peru, (b) per order of class Mammalia (n = 534), (c) per order of class Amphibia (n = 50), (d) per order of class Reptilia (n = 16), (e) per order of class Aves (n = 493), and (f) per order of class Magnoliopsida (n = 76).
Overall, we were able to identify 14 samples only to genus level. Of these, 11 were plants identified in the field, and matching consensus sequences to existing reference databases did not allow for the identification of a single best match; often multiple plant sequences showed >99% identity to our consensus sequence. This implies that these plants require a formal identification, which was not possible with this collection since only imagery was saved and no pressed specimen.
The remaining three samples identified to genus only were mammals: a) one Eptesicus sp. (MUSM 18431) matched with 94.5% identity to a potential new species informally named Eptesicus sp. Peru, FMNH 174918/Genbank accession number OP137039.199 b) two samples of Callicebus sp. (MDPRM017-22) matched Callicebus sp. 2 MH-2016 (KX353795) with 98% similarity based on the cyt b gene. This specimen was described as a different lineage from Calllicebus cupreus and C. toppini100. Further examination of both cyt b and COI in the LACC is warranted due to previously described coloration variation101.
In addition, our data support the first records of Marmosops ocellatus for Peru, a marsupial of the family Didelphidae that is reported presently only with the Amazonian region of Brazil and Bolivia102. We report two specimens housed at the MHN-UNMSM (LT14-18 and MUSM54152) that have COI genetic sequences and one (MUSM54152) that has the cyt b gene sequenced. While the cyt b gene of our specimen matches with 99.5% identity with M. ocellatus in Genbank (OP380991, from Brazil, Acre, Río Blanco)103, there are no recorded COI sequences in any repository, making our COI barcodes the first for this species globally.
We also report the first record of Sturnira lilium for the country of Peru, a bat of the family Phyllostomidae, from specimens collected at the Los Amigos Conservation Concession. We have COI barcodes for two specimens (FPI0007409 and FPI0007439), and cyt b only for FPI0007439. The cyt b sequence matches 100% with Sturnira lilium (genbank accession number: KC753800 /FMNH 162524104 from Tarija, Bolivia and the COI sequence matches 100% with Sturnira lilium (BOLD:AAA1220/Genbank Accession code:OM978245 from Barueri, Brazil). Further study on additional samples and the collection of a voucher is warranted at this site, including considerations of possible mito-nuclear discordance105,106.
Recent barcoding gap analyses in DNA barcoding of birds55 and mammals56 for Peru provide a basis for evaluation of the novel contributions of this dataset for those two groups; a similar evaluation could not be performed for plants, reptiles or amphibians at this time. For mammals, Pacheco et al.56 report that of the 326 Amazonian mammals in Peru, 180 (55.2%) have at least one genetic barcode in a public database. Our study added the first genetic marker for 30 additional species (across 100 barcodes), bringing the total to 210 out of 326 (64.4%), an increase of 9.2%. This includes barcodes of COI for 74 species (raising representation from 11.7% to 22.7% of mammals, a 2 fold increase), cyt b for 26 species (51.2% to 59.2%), and 18S for 32 species of Peruvian mammals (baseline yet unknown) (File 2 available at Figshare59).
Similarly, Amazonian avian species in Peru comprise 1,506 species (79.5% of the 1,894 total avian species reported in Peru)55. Of these, 87.4% (1,316 species) have at least one genetic marker (1239 spp with COI and 897 spp with cyt b), but considering markers generated from Peruvian specimens alone, that figure drops to only 4.3% (n = 64, COI). In this study, we produced the first COI barcodes for 192 additional Peruvian avian species generated from birds within Peru, an increase of 12.7%, contributing to 17.0% of Peruvian Amazonian birds with at least one genetic marker. When considering barcodes generated for birds on Peru’s country list from globally collected specimens, we contributed markers to 65 species (COI and cyt b), increasing numbers by 1.2% (COI) and 6.8% (cyt b). Finally, we generated 38 novel barcodes for 17 species that had no prior genetic reference material, based on the gap analysis55 (File 2 available at Figshare59).
As of July 25, 2024, there were 93,246 sequences from specimens collected in Peru publicly visible on BOLD. Of these, 2,413 were from Aves, 699 were from Mammalia, 352 were from Amphibia, 76 from Reptilia, and 3,873 from Magnoliopsida. With this study, we raise these numbers by 26.55% for Aves (to 3285 barcodes), 52.16% for Mammals (to 1,461 barcodes), 12.7% for Amphibia (to 403 barcodes), 21.7% for Reptilia (to 97 barcodes), and 3.10% for Magnoliopsida (to 3997 barcodes). Overall, this increases the total number of public barcodes for Peru on BOLD by 1.92%, to 95,076 barcodes.
Technical Validation
Blank filtering
Two extraction blanks per batch were included in all DNA extractions, and 1 PCR blank was included in each PCR run. All blanks were taken through to sequencing and subject to the same rigorous filtering criteria applied to all samples. Gel electrophoresis was performed to conduct initial screening for appropriate amplicon sizes after PCR1, and gels after PCR2 were examined to confirm a ~50 bp increase to pools once indices were added.
Sequence quality
Despite early sequencing runs being conducted on earlier chemistries, we saved all raw data and basecalled them again using more accurate algorithms to improve overall read quality scores. Post sequence filtering was conducted (for length -per marker- and quality score cutoff of seven) to eliminate poor quality reads before demultiplexing. Once consensus sequences were formed, any consensus sequences with < 30 supporting reads were discarded. In cases with multiple consensus sequences, the sequence with the closest expected length and the highest coverage was chosen. The sequences of coding genes (cyt b, COI, rbcl, and matk) were visually inspected and manually revised to avoid stop codons using Geneious (v7.1.3 Biomatters Ltd). If a stop codon was detected, we blasted our sequences and checked the potential cause for the stop codon. In 12% of the 1830 sequences a stop codon was due to an extra base pair in a homopolymeric region, a known cause of erroneous bases during sequencing for ONT107; in these cases, we eliminated the extra base.
Laboratory error or contamination
Consensus sequences were meticulously screened using an iterative process described in Fig. 3. To detect potential laboratory errors (e.g., a pipetting error causing sample or index cross-contamination) and correct them, the file containing all consensus sequences was split into higher taxonomic groups per genetic marker. Each file was then aligned in MAFFT version 7 (online platform, https://mafft.cbrc.jp/alignment/server/index.html) using default parameters with the “adjust direction” option enabled. To create a quick and efficient way to find these errors, we used a Neighbor Joining (NJ) tree built with default parameters adjusted to match a Jukes Cantor substitution model of 1,000 bootstrapped iterations. Each tree was then visually inspected in FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/) to ensure that species within the same genus formed a monophyletic group. Any species found nested within an unexpected taxonomic order were discarded. We include detailed examples of this process in File 3: error correction available at Figshare59.
Mismatches of field and genetic IDs
The top reference sequence from GenBank/BOLD was identified as the one with the highest maximum bit score, which correlated with high percent identity and query coverage and low e-values. Mismatches were identified using a cutoff of 95% identity between the consensus and reference sequences. For 340 specimens, one or more barcodes displayed a percent identity > = 95% to a species that was different from the field identification made during a field survey. In these cases of a well-supported mismatch to a different species, we examined the imagery obtained during the survey, checked for newly described species or if the group was subject to a recent taxonomic or systematic revision, and retained the genetic ID if supported by these lines of evidence (n = 327 specimens). In a subset of 189 specimens, field identification beyond order was missing due to collection of the specimen by a non-expert during the field survey, and we were able to successfully determine species identification from DNA barcode results. In cases where percent identification was <95%, we first checked our dataset to see if any other marker with > = 95% identity existed for the same specimen. If this was the case, we would assign these barcodes the same species as this existing marker. If our dataset didn’t contain such a marker, we searched again for any newly published sequences for the species, to eliminate the possibility of a recent taxonomic reclassification. Only in the absence of evidence in this regard, did we conclude that this could be indicative of a potentially new genetic marker for the species. We include detailed examples of this process in File 3: error correction available at Figshare59.
Usage Notes
The recommended workflow for this project is straightforward to repeat and is fully documented for both the laboratory89 and the bioinformatics pipelines90. All processed data and analysis details herein described are made available through open-access resources. Please review the Methods section and online protocols for questions pertaining to data collection, handling, and analysis. If any information seems missing, send an inquiry to the corresponding author.
Code availability
All analyses for this project were carried out using open source software tools. The majority of required software has been packaged into a public Docker image that functions on any operating system that supports Docker. A few additional tools are freely accessible online, such as MAFFT and Posit Cloud for collaborative analyses using R. The full data analysis workflow and associated scripts are available and annotated as a public protocol90. There are no restrictions to access or reproducibility. Software versions remain static within Docker images, and image updates are versioned on Docker Hub.
References
Veach, V., Di Minin, E., Pouzols, F. M. & Moilanen, A. Species richness as criterion for global conservation area placement leads to large losses in coverage of biodiversity. Divers. Distrib. 23, 715–726 (2017).
Pipins, S. et al. Advancing EDGE Zones to identify spatial conservation priorities of tetrapod evolutionary history. Nat. Commun. 15, 7672 (2024).
Pollock, L. J. et al. Protecting biodiversity (in all its complexity): New models and methods. Trends Ecol. Evol. 35, 1119–1128 (2020).
Hopkins, G. W. & Freckleton, R. P. Declines in the numbers of amateur and professional taxonomists: implications for conservation. Animal Conservation forum 5, 245–249 (2002).
Scotland, R. W., Olmstead, R. G. & Bennett, J. R. Phylogeny reconstruction: the role of morphology. Syst. Biol. 52, 539–548 (2003).
Engel, M. S. et al. The taxonomic impediment: a shortage of taxonomists, not the lack of technical approaches. Zool. J. Linn. Soc. 193, 381–387 (2021).
Hanson, T. et al. Warfare in biodiversity hotspots. Conserv. Biol. 23, 578–587 (2009).
Grooten, M. & Almond, R. E. A. Living Planet Report 2018: Aiming Higher: Summary. (2018).
Proença, V. et al. Global biodiversity monitoring: From data sources to essential biodiversity variables. Biol. Cons. 213, 256–263, https://doi.org/10.1016/j.biocon.2016.07.014 (2017).
Pereira, H. M., Navarro, L. M. & Martins, I. S. Global biodiversity change: The bad, the good, and the unknown. Annu. Rev. Environ. Resour. 37, 25–50, https://doi.org/10.1146/annurev-environ-042911-093511 (2012).
Herman, S. G. Wildlife biology and natural history: Time for a reunion. J. Wildl. Manag. 66, 933-946, https://doi.org/10.2307/3802927 (2002).
Schmidly, D. J. What it means to be a naturalist and the future of natural history at american universities. J. Mammal. 86, 449–456, https://doi.org/10.1644/1545-1542(2005)86[449:wimtba]2.0.co;2 (2005).
Greene, H. W. Organisms in nature as a central focus for biology. Trends Ecol. Evol. 20, 23–27 (2005).
Mora, C., Tittensor, D. P., Adl, S., Simpson, A. G. B. & Worm, B. How many species are there on Earth and in the ocean? PLoS Biol. 9, e1001127, https://doi.org/10.1371/journal.pbio.1001127 (2011).
Fisher, M. A., Vinson, J. E., Gittleman, J. L. & Drake, J. M. The description and number of undiscovered mammal species. Ecol. Evol. 8, 3628–3635 (2018).
Hebert, P. D. N. & Gregory, T. R. The promise of DNA barcoding for taxonomy. Syst. Biol. 54, 852–859 (2005).
Pomerantz, A. et al. Real-time DNA barcoding in a rainforest using nanopore sequencing: opportunities for rapid biodiversity assessments and local capacity building. Gigascience 7 1-14 (2018).
Krehenwinkel, H., Pomerantz, A. & Prost, S. Genetic biomonitoring and biodiversity assessment using portable sequencing technologies: Current uses and future directions. Genes 10, 1–16 (2019).
Watsa, M., Erkenswick, G., Pomerantz, A. & Prost, S. Portable sequencing as a teaching tool in conservation and biodiversity research. PLoS Biol. 18, e3000667 (2020).
Menegon, M. et al. On site DNA barcoding by nanopore sequencing. PLoS One 12, e0184741 (2017).
Krehenwinkel, H. et al. Nanopore sequencing of long ribosomal DNA amplicons enables portable and simple biodiversity assessments with high phylogenetic resolution across broad taxonomic scale. Gigascience 8, 1–16 (2019).
Srivathsan, A. et al. Rapid, large-scale species discovery in hyperdiverse taxa using 1D MinION sequencing. BMC Biol. 17, 96 (2019).
DeSalle, R. & Goldstein, P. Review and interpretation of trends in DNA barcoding. Front. Ecol. Evol. 7, 302 (2019).
Gaston, K. J. & O’Neill, M. A. Automated species identification: why not? Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 655–667 (2004).
Hebert, P. D. N., Ratnasingham, S. & deWaard, J. R. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc. Biol. Sci. 270(Suppl 1), S96–9 (2003).
Ivanova, N. V., Borisenko, A. V. & Hebert, P. D. N. Express barcodes: racing from specimen to identification. Mol. Ecol. Resour. 9(Suppl s1), 35–41 (2009).
Mena, J. L. et al. Environmental DNA metabarcoding as a useful tool for evaluating terrestrial mammal diversity in tropical forests. Ecol. Appl. 31, e02335 (2021).
Valentini, A., Pompanon, F. & Taberlet, P. DNA barcoding for ecologists. Trends Ecol. Evol. 24, 110–117 (2009).
Janzen, D. & Hallwachs, W. How a tropical country can DNA barcode itself. iBOL Barcode Bulletin 9, 1–6, https://doi.org/10.21083/ibol.v9i1.5526 (2019).
Lorenz, J. G., Jackson, W. E., Beck, J. C. & Hanner, R. The problems and promise of DNA barcodes for species diagnosis of primate biomaterials. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360, 1869–1877 (2005).
Yahr, R., Schoch, C. L. & Dentinger, B. T. M. Scaling up discovery of hidden diversity in fungi: impacts of barcoding approaches. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371 (2016).
Ratnasingham, S. et al. BOLD v4: A centralized bioinformatics platform for DNA-based biodiversity data. Methods Mol. Biol. 2744, 403–441 (2024).
Jenkins, C. N., Pimm, S. L. & Joppa, L. N. Global patterns of terrestrial vertebrate diversity and conservation. Proc. Natl. Acad. Sci. USA. 110, E2602–10 (2013).
Urban, L. et al. Real-time genomics for One Health. Mol. Syst. Biol. 19, e11686 (2023).
Bertola, L. D. et al. A pragmatic approach for integrating molecular tools into biodiversity conservation. Conserv. Sci. Pract. 6 (2024).
Heather, J. M. & Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 107, 1–8 (2016).
Sanger, F., Nicklen, S. & Coulson, A. R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74, 5463–5467 (1977).
Prosser, S. W. J., deWaard, J. R., Miller, S. E. & Hebert, P. D. N. DNA barcodes from century-old type specimens using next-generation sequencing. Mol. Ecol. Resour. 16, 487–497 (2016).
Shokralla, S. et al. Next-generation DNA barcoding: using next-generation sequencing to enhance and accelerate DNA barcode capture from single specimens. Mol. Ecol. Resour. 14, 892–901 (2014).
Pomerantz, A. et al. Rapid in situ identification of biological specimens via DNA amplicon sequencing using miniaturized laboratory equipment. Nat. Protoc. 17, 1415–1443 (2022).
Hole, K. & Nfon, C. Foot‐and‐mouth disease virus detection on a handheld real‐time polymerase chain reaction platform. Transbound. Emerg. Dis. 129, 268 (2019).
Srivathsan, A. et al. A MinIONTM-based pipeline for fast and cost-effective DNA barcoding. Mol. Ecol. Resour. 18, 1035–1049 (2018).
Gallagher, M. D. et al. Nanopore sequencing for rapid diagnostics of salmonid RNA viruses. Sci. Rep. 8, 16307 (2018).
Truelove, N. K., Andruszkiewicz, E. A. & Block, B. A. A rapid environmental DNA method for detecting white sharks in the open ocean. Methods Ecol. Evol. 10, 1128–1135 (2019).
Latorre-Pérez, A. et al. A Round trip to the desert: In situ nanopore sequencing informs targeted bioprospecting. Front. Microbiol. 12, 768240 (2021).
De Vivo, M. et al. Utilisation of Oxford Nanopore sequencing to generate six complete gastropod mitochondrial genomes as part of a biodiversity curriculum. Sci. Rep. 12, 9973 (2022).
Prost, S. et al. Improving the chromosome-level genome assembly of the Siamese fighting fish (Betta splendens) in a University Master's course. G3 10, 2179–2183 (2020).
Lin, X., Waring, K., Ghezzi, H., Tropini, C., Tyson, J. & Ziels, R. M. High-accuracy meets high-throughput for microbiome profiling with near full-length 16S rRNA amplicon sequencing on the Nanopore platform. PNAS Nexus 3(10), https://doi.org/10.1093/pnasnexus/pgae411 (2023).
Plenge, M. A. List of the birds of Peru/ Lista de las aves del Peru. Lista de las aves del Peru (2024).
The Reptile Database: Curating the biodiversity literature without funding. 246–255 (2021).
Uetz, P. et al. A quarter century of reptile and amphibian databases. Herpetologica.
Froese, R. & Pauly, D. FishBase 2000: Concepts Designs and Data Sources. (WorldFish, 2000).
Secretariat for the Convention on Biological Diversity. Peru-Country Profile. Convention on Biological Diversity https://www.cbd.int/countries/profile?country=pe.
Pacheco, V. et al. Lista actualizada de la diversidad de los mamíferos del Perú y una propuesta para su actualización. Rev. Peru. Biol. 28, e21019 (2021).
Arana, A. et al. Lack of local genetic representation in one of the regions with the highest bird species richness, the Peruvian Amazonia. PLoS One 19, e0296305 (2024).
Pacheco, V. et al. Disproportion between the Peruvian Amazonian megadiverse mammalian fauna and the available molecular information. Zoologia 41, e23110 (2024).
Kress, W. J. Plant DNA barcodes: Applications today and in the future. J. Syst. Evol. 55, 291–307 (2017).
Lima, R. A. F. de et al. Can plant DNA barcoding be implemented in species-rich tropical regions? A perspective from São Paulo State, Brazil. Genet. Mol. Biol. 41, 661–670 (2018).
Sánchez-Vendizú, P. et al. Decoding the Peruvian Amazon with in situ DNA barcoding of vertebrate and plant taxa, Figshare., https://doi.org/10.6084/m9.figshare.29242538 (2025).
Britto, B. Actualización de las Ecorregiones Terrestres de Perú propuestas en el Libro Rojo de Plantas Endémicas del Perú. Gayana Bot. 74, 15–29 (2017).
MINAM. Reporte: Madre de Dios: Estadísticas Ambientales. https://sinia.minam.gob.pe/documentos/madre-dios-estadisticas-ambientales-diciembre-2019 (2019).
Longmire, J. L., Albright, K. L., Lewis, A. K., Meincke, L. J. & Hildebrand, C. E. A rapid and simple method for the isolation of high molecular weight cellular and chromosome-specific DNA in solution without the use of organic solvents. Nucleic Acids Res. 15, 859 (1987).
Huang, Y.-T. RNAlater Recipe. https://www.protocols.io/view/rnalater-recipe-ccm4su8w, https://doi.org/10.17504/protocols.io.bp2l61w35vqe/v1 (2022).
Mendoza-Silva, J. L. et al. Handling and Sampling Birds - ISL Peru. https://www.protocols.io/view/handling-and-sampling-birds-isl-peru-cr9iv94e, https://doi.org/10.17504/protocols.io.6qpvr4zwpgmk/v1 (2023).
Watsa, M. et al. Handling and Sampling Bats - ISL Peru. 1, https://www.protocols.io/view/handling-and-sampling-bats-isl-peru-q26g7y7o9gwz/v1, https://doi.org/10.17504/protocols.io.q26g7y7o9gwz/v1 (2023).
Tirapelle, C. et al. Handling and Sampling Small Non-Volant Mammals - ISL Peru. https://doi.org/10.17504/protocols.io.kxygx9xkdg8j/v1 (2023).
Watsa, M. et al. Handling and Sampling Small Nonhuman Primates - ISL Peru. https://www.protocols.io/view/handling-and-sampling-small-nonhuman-primates-isl-crtqv6mw, https://doi.org/10.17504/protocols.io.5qpvor1exv4o/v1 (2023).
Tirapelle, C., Mendoza-Silva, J. L., Watsa, M. & Erkenswick, G. Handling and Sampling Medium-Large Mammals - ISL Peru. https://www.protocols.io/view/handling-and-sampling-medium-large-mammals-isl-per-cryzv7x6 (2023).
Libke, Z., Paine, T., Erkenswick, G. & Watsa, M. Handling and Sampling Herpetofauna - ISL Peru. https://www.protocols.io/view/handling-and-sampling-herpetofauna-isl-peru-crt8v6rw (2023).
Guidelines for the treatment of animals in behavioural research and teaching. Anim. Behav. 71, 245–253 https://doi.org/10.1016/j.anbehav.2005.10.001 (2006).
Sikes, R. S., Animal Care and Use Committee of the American Society of Mammalogists. 2016 Guidelines of the American Society of Mammalogists for the use of wild mammals in research and education. J. Mammal. 97, 663–688 (2016).
Kress, W. J. & Erickson, D. L. DNA barcodes: methods and protocols. Methods Mol. Biol. 858, 3–8 (2012).
De Volo, S. B., Reynolds, R. T., Douglas, M. R. & Antolin, M. F. An improved extraction method to increase DNA yield from molted feathers. Condor 110, 762–766 (2008).
Vasquez, J. R., Sánchez-Vendizú, P., Vsevolodova, K. N, Erkenswick, G., Watsa, M. ISL Opentrons Pipeline: gDNA Bead Cleanup. https://doi.org/10.17504/protocols.io.3byl4j9pzlo5/v2 (2023).
Chambers, E. A. & Hebert, P. D. N. Assessing DNA barcodes for species identification in North American reptiles and amphibians in natural history collections. PLoS One 11, e0154363 (2016).
Naidu, A., Fitak, R. R., Munguia-Vega, A. & Culver, M. Novel primers for complete mitochondrial cytochrome b gene sequencing in mammals. Mol. Ecol. Resour. 12, 191 (2012).
Smith, M. F. & Patton, J. L. The diversification of South American murid rodents: evidence from mitochondrial DNA sequence data for the akodontine tribe. Biol. J. Linn. Soc. Lond. (1993).
Hebert, P. D. N., Stoeckle, M. Y., Zemlak, T. S. & Francis, C. M. Identification of birds through DNA barcodes. PLoS Biol. 2, e312 (2004).
Kerr, K. C. R., Lijtmaer, D. A., Barreira, A. S., Hebert, P. D. N. & Tubaro, P. L. Probing evolutionary patterns in neotropical birds through DNA barcodes. PLoS One 4, e4379 (2009).
Johnson, J. A., Lerner, H. R. L., Rasmussen, P. C. & Mindell, D. P. Systematics within Gyps vultures: a clade at risk. BMC Evol. Biol. 6, 65 (2006).
Holmgaard, D. B. et al. Detection and identification of Acanthamoeba and other nonviral causes of infectious keratitis in corneal scrapings by real-time PCR and next-generation sequencing-based 16S–18S gene analysis. J. Clin. Microbiol. 59, https://doi.org/10.1128/JCM.02224-20 (2021).
Cuénoud, P. et al. Molecular phylogenetics of Caryophyllales based on nuclear 18S rDNA and plastid rbcL, atpB, and matK DNA sequences. Am. J. Bot. 89, 132–144 (2002).
Tate, J. A. & Simpson, B. B. Paraphyly of Tarasa (Malvaceae) and diverse origins of the polyploid species. Syst. Bot. 28, 723–737 (2003).
Sang, T., Crawford, D. & Stuessy, T. Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Am. J. Bot. 84, 1120 (1997).
Levin, R. A. et al. Family-level relationships of Onagraceae based on chloroplast rbcL and ndhF data. Am. J. Bot. 90, 107–115 (2003).
Kress, W. J. & Erickson, D. L. A two-locus global DNA barcode for land plants: the coding rbcL gene complements the non-coding trnH-psbA spacer region. PLoS One 2, e508 (2007).
Vasquez, J. R. et al. Opentrons Pipeline: PCR Preparation. https://doi.org/10.17504/protocols.io.rm7vzx6y8gx1/v1 (2023).
Jolivet, P. & Foley, J. W. Solutions for purifying nucleic acids by solidphase reversible immobilization (SPRI). Ludmer Center, Neuroinfromatics & Mental Health 1–6 (2015).
Watsa, M., Vsevolodova, K., Sánchez-Vendizú, P. & Erkenswick, G. Custom indexing for nanopore sequencing platform. Protocols.io https://doi.org/10.17504/protocols.io.q26g71ed8gwz/v1 (2024).
Erkenswick, G., Watsa, M., Libke, Z., Sánchez-Vendizú, P. & Prost, S. Species molecular barcode analysis with nanopore sequence data. Protocols.io https://doi.org/10.17504/protocols.io.6qpvr8y1blmk/v1 (2024).
De Coster, W. & Rademakers, R. NanoPack2: population-scale evaluation of long-read sequencing data. Bioinformatics 39 (2023).
Sahlin, K., Lim, M. C. W. & Prost, S. NGSpeciesID: DNA barcode and amplicon consensus generation from long-read sequencing data. Ecol. Evol. 11, 1392–1398 (2021).
Sahlin, K. & Medvedev, P. De novo clustering of long-read transcriptome data using a greedy, quality value-based algorithm. J. Comput. Biol. 27, 472–484 (2020).
Ogden, R., Vasiljevic, N. & Prost, S. Nanopore sequencing in non-human forensic genetics. Emerg Top Life Sci 5, 465–473 (2021).
Vasiljevic, N. et al. Developmental validation of Oxford Nanopore Technology MinION sequence data and the NGSpeciesID bioinformatic pipeline for forensic genetic species identification. Forensic Sci. Int. Genet. 53, 102493 (2021).
Hebert, P. D. N., Floyd, R., Jafarpour, S. & Prosser, S. W. J. Barcode 100K specimens: in a single nanopore run. Mol. Ecol. Resour. 25, e14028 (2025)
Zhang, T. et al. The newest Oxford Nanopore R10.4.1 full-length 16S rRNA sequencing enables the accurate resolution of species-level microbial community profiling. Appl. Environ. Microbiol. 89, e0060523 (2023).
DS-ISLPE24. DNA Barcodes from In Situ Laboratories in Peru. Bold. https://doi.org/10.5883/DS-ISLPE24 (2024).
Yi, X. et al. UCE-derived mitochondrial phylogeny reveals pervasive mito-nuclear discordances in serotine bats (genus Eptesicus) and complex evolutionary history in Eptesicus (Histiotus). Mamm. Biol. https://doi.org/10.1007/s42991-024-00418-2 (2024).
Hoyos, M. et al. Phylogenetic relationships within the Callicebus cupreus species group (Pitheciidae: Primates): Biogeographic and taxonomic implications. Mol. Phylogenet. Evol. 102, 208–219 (2016).
Vermeer, J. & Tello-Alvarado, J. C. The Distribution and taxonomy of Titi monkeys (Callicebus) in Central and Southern Peru, with the description of a new species. Prim. Cons. 9–29 (2015).
Voss, R. S., Tarifa, T. & Yensen, E. An introduction to Marmosops (marsupialia: Didelphidae), with the description of a new species from Bolivia and notes on the taxonomy and distribution of other Bolivian forms. Am. Mus. Novit. 3466, 1–40 (2004).
Bonvicino, C. R. et al. Phylogeny of Didelphid marsupials (Didelphimorphia) from Acre, western Amazonia. Mamm. Biol. 102, 1997–2015 (2022).
Velazco, P. M. & Patterson, B. D. Diversification of the yellow-shouldered bats, genus Sturnira (Chiroptera, Phyllostomidae), in the New World tropics. Mol. Phylogenet. Evol. 68, 683–698 (2013).
Mao, X. et al. Multiple cases of asymmetric introgression among horseshoe bats detected by phylogenetic conflicts across loci. Biol. J. Linn. Soc. Lond. 110, 346–361 (2013).
Dávalos, L. M., Cirranello, A. L., Geisler, J. H. & Simmons, N. B. Understanding phylogenetic incongruence: lessons from phyllostomid bats. Biol. Rev. Camb. Philos. Soc. 87, 991–1024 (2012).
Huang, Y.-T., Liu, P.-Y. & Shih, P.-W. Homopolish: a method for the removal of systematic errors in nanopore sequencing by homologous polishing. Genome Biol. 22, 95 (2021).
Acknowledgements
We would like to thank the teams of the Museo de Historial Natural of the Universidad Nacional Mayor de San Marcos (UNMSM), Fheld Projects International, Conservación-Amazónica-ACCA, Taricaya Rehabilitation Center, Amazon Shelter and the Inkaterra Association for their invaluable time spent collecting, cataloging and barcoding the biospecimens utilized in this effort. The work at the UNMSe was supported by PROCIENCIA/Concytec, Project Code: 039-2021. LS and CA were funded by VRIP-UNMSM, Project codes: B24100351, B23100441 and B22100321. VP was funded by CONCYTEC through the PROCIENCIA program within the framework of the ‘Basic Research Projects’ contest 2022-01, according to contract N° PE501078825-2022-PROCIENCIA. SP is funded by the University of Oulu and the Academy of Finland Profi6 336449 programme “Biodiverse Anthropocenes”. The GL was supported by the Inkaterra Foundation. The WCL-Los Amigos was supported by subawards from the Gordon and Betty Moore Foundation (grants 9776 and 9772), the San Diego Zoo Wildlife Alliance, Field Projects International, and the Amazon Conservation Association.
Author information
Authors and Affiliations
Contributions
P.S.V.: Writing - Original Draft, Data Collection, Taxonomic identification, Laboratory Analysis, Formal Analysis. G.E.: Conceptualization, Methodology, Data Collection, Laboratory Analysis, Funding Acquisition, Laboratory Installation, Formal Analysis, Bioinformatic Analysis, Writing - Original Draft, Supervision. J.R.: Methodology, Laboratory Analysis, Formal Analysis, Writing - Review & Editing. S.L.C.: Data Collection - field surveys, Laboratory Installation, Laboratory Analysis, Writing - Review & Editing. T.S.E.: Methodology, Laboratory Analysis, Formal Analysis, Writing - Review & Editing. G.C.: Methodology, Laboratory Analysis, Formal Analysis. Z.L.: Data collection - taxonomic identification, Laboratory Analysis, Formal Analysis, Writing - Review & Editing. A.A.: Data collection, Laboratory Analysis, Taxonomic identification, Formal Analysis, Writing - Review & Editing. J.M.S.: Data Collection - taxonomic identification, Writing - Review & Editing. C.T.: Data Collection, Writing - Review & Editing. S.W.: Data Collection - taxonomic identification, Writing - Review & Editing. V.S.: Conceptualization, Methodology, Data Collection - field surveys, Formal Analysis, Writing - Review & Editing, Funding Acquisition. J.M.A.: Data collection, Laboratory Analysis, Writing - Review & Editing. J.E.: Data collection, Laboratory Analysis, Writing - Review & Editing. J.P.B.: Laboratory Analysis, Writing - Review & Editing. J.H.M.: Data collection, Laboratory Analysis, Writing - Review & Editing. X.C.: Data collection, Laboratory Analysis, Formal Analysis, Writing - Review & Editing. A.M.M.: Data collection, Laboratory Analysis, Writing - Review & Editing. R.S.A.: Data Collection, Writing - Review & Editing. L.G.J.: Data Collection, Writing - Review & Editing. J.S.: Data Collection - taxonomic identification, Writing - Review & Editing. N.C.: Data Collection, Writing - Review & Editing. T.D.P.: Data Collection - taxonomic identification, Writing - Review & Editing. G.I.D.: Data Collection - taxonomic identification, Writing - Review & Editing. P.P.A.: Data Collection, Writing - Review & Editing. J.L.: Data Collection, Writing - Review & Editing. A.V.M.: Sample collection - field surveys, Laboratory Analysis, Review - Draft. M.E.M.: Laboratory Analysis, Writing - Review & Editing D.C.: Data Collection, Writing - Review & Editing. L.L.R.: Laboratory Analysis, Writing - Review & Editing N.W.P.: Laboratory Analysis, Writing - Reviewing & Editing A.J.S.: Data Collection - field surveys, Writing - Reviewing & Editing M.A.: Data collection, Writing - Review & Editing. J.L.M.: Ceonceptualization, Funding Acquisition, Supervision. M.W.T.: Conceptualization, Funding Acquisition, Supervision. L.S.: Data collection, Writing - Review & Editing, Taxonomic identification, Funding Acquisition, Supervision. C.A.: Data collection, Writing - Review & Editing, Taxonomic identification, Funding Acquisition, Supervision. V.P.: Data collection, Writing - Review & Editing, Taxonomic identification, Funding Acquisition, Supervision. S.P.: Conceptualization, Methodology, Laboratory Analysis, Bioinformatic Analysis, Writing - Draft, Funding Acquisition, Supervision. M.W.: Conceptualization, Methodology, Data Collection - field surveys, Laboratory Analysis, Formal Analysis, Bioinformatic Analysis, Writing - Original Draft, Funding Acquisition, Supervision.
Corresponding author
Ethics declarations
Competing interests
M.W. and Z.L have received travel and lodging reimbursement from Oxford Nanopore Technologies (ONT) as invited keynote speakers. Z.L., M.W., S.P., G.E. and the In Situ Laboratory Network have also received donated consumables and sequencers from ONT as beta users of the P2solo and MK1d devices.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sánchez-Vendizú, P., Erkenswick, G., Reyes, J. et al. Decoding the Peruvian Amazon with in situ DNA barcoding of vertebrate and plant taxa. Sci Data 12, 1545 (2025). https://doi.org/10.1038/s41597-025-05697-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05697-z
