
Abstract
Hu sheep is an indigenous breed in China, renowned for its prolificacy. However, Hu sheep have smaller statures compared to other meat sheep breeds, necessitating improvement. Therefore, further research is required to explore the underlying molecular genetic mechanisms of body size traits in Hu sheep. In this study, whole genome sequencing was conducted on 300 Hu sheep with an average depth of 16.51X. A total of 9.53 T of high-quality sequencing data was generated. After quality controlled, Q30 range of clean reads was 86.76% to 95.46%, and GC range was 41.52% to 44.48%. Subsequently, we identified 23274312 single nucleotide polymorphisms (SNPs) and 64759 structural variations (SVs) through a series of bioinformatics analyses. Genome-wide association studies (GWAS), including SNP-GWAS and SV-GWAS, were performed in combination with five body size traits. Furthermore, domestication adaptation regions within the Hu sheep population were explored through selection signatures analysis. This dataset provides a valuable genetic resource for sheep breeding, and serves as a reference for the application of SVs in sheep economic traits.
Similar content being viewed by others

Background & Summary
Sheep body size is closely correlated with meat production1, fat deposition2, and reproductive performance3. The consumption of meat continues to increase globally; the larger body size will bring higher profits to sheep farmers4. Hu sheep have excellent reproductive performance. It has many other advantages, such as early fast growth and development, low fatty content and high carcass yield5. In recent years, because of the increase demand for mutton and the decrease demand for sheepskin, the Hu sheep industry needs to be upgraded6. However, Hu sheep have smaller statures compared to other meat sheep breeds, necessitating improvement. Sheep body size can be influenced by various factors, especially genetics7. Whole genome sequencing provides a large number of genetic variations, which helps to further explore the underlying molecular genetic mechanisms of body size traits in Hu sheep.
Most studies on economic traits in sheep had focused only on SNPs and neglected SVs. Compared to SNPs, SVs have a more direct impact on phenotype, and can explain more complex genetic variations8. Currently, researches on sheep SV are mainly focused on evolution and development9,10,11, and few studies have systematically analyzed the effects of SV on economic traits in sheep. The field is largely unexplored. Therefore, the effects of SV on body size traits in Hu sheep need to be further investigated. Quantitative trait loci (QTL) and selection signatures have been widely used in the study of livestock traits. The QTL, GWAS and selection signatures analyses complement each other to identify candidate loci more accurately12. In a previous study, we explored the evolutionary history of Hu sheep in conjunction with other Mongolian sheep breeds6.
In this study, we conducted whole genome sequencing on 300 Hu sheep with an average sequencing depth of 16.51X. A total of 9.53 T high-quality data was obtained. Five body size traits were recorded, including body weight (BW), body length (BL), body height (BH), chest circumference (CC) and cannon bone circumference (CBC). Combined with these body size traits, GWAS based on SNPs and SVs were performed, respectively. Furthermore, we had analyzed the selection signatures of SNPs and SVs in Hu sheep population. This dataset contributes to a more comprehensive understanding of genetic variations in Hu sheep, and provides new perspectives on the conservation of Hu sheep genetic resources.
Methods
Ethics statement
The animal study protocol was reviewed and approved by the Institutional Animal Care and Use Committee of Zhejiang University (ZJU25331).
Hu sheep samples collection
In this study, all Hu sheep (n = 300) utilized were collected from the Yihui Ecological Agriculture Co, Huzhou City, Zhejiang Province, China. All Hu sheep were raised in the same conditions. Each phenotype was measured by the same person to minimize measurement error. BW was measured by specific electronic scale. BL was the straight-line distance from the anterior end of the scapula to the posterior end of the sciatic tuberosity; BH was the vertical distance from the highest point of the withers to the ground surface; CC referred to girth measurement of the posterior end of the scapula around thorax; CBC was the circumference of the tibial third of the left forelimb. Table 1 demonstrated the statistical data for above five traits. Two milliliters of blood samples were collected for DNA extraction.
Library construction and sequencing
The magnetic bead method was utilized to extract DNA, and the DNA samples were tested for integrity and purity before being accurately quantified. Only DNA samples that passed the test could be used for library construction. After library construction was completed and passed the test, qualified samples were sequenced. The raw data would be used in the next step of analysis.
Identification of SNPs
Before conducting bioinformatics analysis, the raw data required to be filtered and quality controlled. Using fastp13 for quality control of raw reads. According to the reference genome ARS-UI_Lamb_v2.014 (https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_016772045.1/), the raw data were filtered for clean data and index. The BWA15 and samtools16 softwares were used to process clean reads and index, respectively, to obtain Bam file. Duplicates were removed from the Bam file using GATK software17. Genome coverage and sequencing depth were calculated for all samples based on the Bam file. Subsequently, the SNPs were filtered using PLINK18, the code was: PLINK --allow-extra-chr --bfile test --chr 1-26 --chr-set 95 --maf 0.05 --geno 0.1 --hwe 0.000001 --out vcf--recode vcf-iid --snps-only just-acgt. Beagle19 was used to fill the missing SNPs.
Identification of SVs
Six software were used to identify SVs. They were Delly (1.2.6)20, Dysgu (1.6.2)21, GRIDSS2 (2.13.2)22, Manta (1.6.0)23, Wham (1.8.0)24, and Smoove (0.2.8)25. All insertions (INS) and deletions (DEL) identified were clustered and combined. For accuracy of identification, only SVs that supported by more than three software were retained. Only INSs with definite sequences were retained according to the breakpoint records of SVs. A set of candidate SVs was composed of the identified INSs and DELs. Based on the SVs candidate set, a pan-genome graph was created, and SV genotyping was performed on all samples with GraphTyper226 software. Subsequently, further filtration was performed on all SVs: MAF > 0.01 and missing rates < 0.3. Beagle19 was used for genotype phasing of SVs with the default parameters.
GWAS based on SNPs and SVs
The rMVP27 software was used for GWAS. The mixed linear model27 accurately explained kinship and population structure. Therefore, we used this model to conduct GWAS. The formula for this model is as follows:
where \(y\) is the phenotype vector, \(X\beta \) is the fixed effects, including population structure, sex, birth year and season, and measurement age of Hu sheep, \({Z}_{k}{\gamma }_{k}\) is the marker effect to be tested, \(\xi \sim N\left(0,K{{\rm{\varnothing }}}^{2}\right)\) represents the polygenic effect, and \(e \sim N\left(0,I{\sigma }^{2}\right)\) is the residual effect. \(K\) is the polygenic effect in the marker-inferred kinship matrix. Manhattan and Q-Q plots were made using CMplot software27. The 1000 permutation test was used to determine the threshold of SNP-GWAS. The threshold of SV-GWAS was determined using the top 5% of −log10(p-value)10.
The Animal QTL Database28 (http://www.animalgenome.org/QTLdb) was used for QTL annotation. The QTL enrichment and gene annotation were performed using the GALLO R package29. Functional annotation of candidate genes was performed in the Herbivore Transcriptome Information Resource Database30 (https://yanglab.hzau.edu.cn/HTIRDB#/).
Selection signature analysis
Integrated haplotype score (IHS) analysis was conducted using selscan software31. PLINK was used for runs of homozygosity (ROH) detection. The following parameters were used for the SNP ROH: PLINK --homozyg-window-threshold 0.05 --homozyg-het 1 --homozyg-window-missing 5 --homozyg-snp 50 --homozyg-kb 500 --homozyg-window-het 1 --homozyg-gap 100 --homozyg-density 50 --homozyg-window-snp 50; the following parameters were used for the SV ROH: PLINK --homozyg-window-missing 5 --homozyg-snp 25 --homozyg --homozyg-gap 1000 --homozyg-het 1 --homozyg-density 150 --homozyg-window-threshold 0.05 --homozyg-window-snp 25 --homozyg-window-het 1 --homozyg-kb 1000. Runs of heterozygosity (ROHet) were identified and analyzed using the detectRUNS R package32. SNP ROHet used the following parameters: minSNP = 10, maxGap = 10^6, minLengthBps = 50000, maxOppRun = 3, maxMissRun = 2; SV ROHet used the following parameters: minSNP = 10, maxGap = 10^6, minLengthBps = 200000, maxOppRun = 3, maxMissRun = 2. The top 0.1% of SNP or SV occurrences were used as the hotspot regions for selection signatures.
Data Records
The raw data used in this study are available in the NGDC database under GSA accession number CRA01783233 (https://ngdc.cncb.ac.cn/gsa/browse/CRA017832). The SNP-VCF and SV-VCF files for this study have been deposited in the European Variation Archive (EVA) at EMBL-EBI under accession number PRJEB9432834.
Technical Validation
Quality control of genomic data
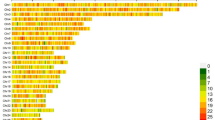
The average sequencing depth of all sample was 16.51X. The average mapping rate of the sequence reads against the reference genome was 97.66%. The Q30 range of clean reads was 86.76% to 95.46%, and GC range was 41.52% to 44.48% (Fig. 1, Table S1). These indicators further confirmed the high quality of the sequencing data.

Boxplot showing the sequencing depth, Q30 content and GC content of Hu sheep samples (n = 300).
Quality control of SNPs and SVs
Following quality control, 23274312 SNPs were obtained from 26 autosomes. The high-density of SNP in 1 mbyte (Mb) was shown in Fig. 2. Furthermore, 64759 SVs were obtained, including 42160 DELs and 22599 INSs (Fig. 3a,b). All SVs were greater than 50 bp, with the largest SV was 62,337 bp DEL.

The number of SNPs within 1MB window size.

The number and type of SVs. The number of SVs within 1MB window size (a). The type of SVs (b).
Code availability
The following code contains the complete process from environment configuration to SV vcf file construction.
1. Environment:
conda create -n SV
source ${CondaPATH}/bin/activate SV
conda install samtools fastp bwa dysgu delly gridss smoove wham graphtyper bcftools vcftools plink SURVIVOR bedtools
2. Mapping and quaqlity control
REF = ${RefferncePATH}/Sheep.fa
samtools faidx $REF
Thread = ${TreadNumber}
source ${CondaPATH}/bin/activate SV
OutDir = ${MissionPATH}/fastq
mkdir $OutDir
cd $OutDir
find ${FastqPATH} -type f -name “*.R1.fastq.gz” | awk -F’.R1.fastq.gz’ ‘{print $1}’ | sort > ${MissionPATH}/IndividualList.txt
while read ID || [[-n ${ID}]]
do
cd $OutDir
mkdir -p OUT/${ID}
RunOUT = ${MissionPATH}/fastq/OUT/${ID}
fastp -i ${FastqPATH}/${ID}.R1.fastq.gz -I ${FastqPATH}/${ID}.R2.fastq.gz -o ${RunOUT}/$ID\_1.QC.fastq.gz -O ${RunOUT}/$ID\_2.QC.fastq.gz -j ${RunOUT}/$ID.fastp.json -h ${RunOUT}/$ID.fastp.html -q 20 -u 30 -l 75 -w $Thread
Fastq1 = ${RunOUT}/$ID\_1.QC.fastq.gz
Fastq2 = ${RunOUT}/$ID\_2.QC.fastq.gz
3. Bam:
export SENTIEON_LICENSE = ${SentieonLicense}
sentieon = ${SentieonPATH}/sentieon
$sentieon bwa mem -t $Thread -R “@RG\tID:$ID\tLB:$ID\tPL:ILLUMINA\tSM:$ID” -M ${MissionPATH}/fastq/REF/Sheep $Fastq1 $Fastq2 | $sentieon util sort -r ${REF} -o $RunOUT/$ID.bam -t $Thread --sam2bam -i -
rm $Fastq1
rm $Fastq2
$sentieon driver -t $Thread --temp_dir $OutDir/TMP/ -i $RunOUT/$ID.bam --algo LocusCollector --fun score_info $RunOUT/$ID.score.txt
$sentieon driver -t $Thread --temp_dir $OutDir/TMP/ -i $RunOUT/$ID.bam --algo Dedup--rmdup --score_info $RunOUT/$ID.score.txt --metrics $RunOUT/$ID.rmdup_metrics.txt $RunOUT/$ID.rmdup.bam
rm $RunOUT/$ID.bam
$sentieon driver -r ${REF} -t $Thread --temp_dir $OutDir/TMP/ -i $RunOUT/$ID.rmdup.bam --algo Realigner $RunOUT/$ID.realigner.bam
rm $RunOUT/$ID.rmdup.bam
done < ${MissionPATH}/IndividualList.txt
4. Index:
find ${MissionPATH}/fastq/OUT/*/*.realigner.bam | sort > ${MissionPATH}/BamList.txt
DIR = ${MissionPATH}/fastq/OUT/${ID}
cd ${DIR}
while read ID || [[-n ${ID}]]
do
Bam = $ID.realigner.bam
samtools index -b $Bam
echo ${ID}
done < ${MissionPATH}/BamList.txt
5. SVcalling (delly):
Ref = ${RefferncePATH}/Sheep.fa
DIR = ${MissionPATH}/Delly
mkdir ${DIR}
cd ${DIR}
source ${CondaPATH}/bin/activate SV
while read ID || [[-n ${ID}]]
do
Bam = ${MissionPATH}/fastq/OUT/${ID}/$ID.realigner.bam
mkdir ${ID}
cd ${ID}
delly call -o ${DIR}/${ID}/${ID}.bcf -g $Ref $Bam
bcftools view ${DIR}/${ID}/${ID}.bcf > ${DIR}/${ID}.Delly.vcf
cd ${DIR}
done < ${MissionPATH}/IndividualList.txt
6. SVcalling (dysgu)
Thread = ${TreadNumber}
Ref = ${RefferncePATH}/Sheep.fa
DIR = ${MissionPATH}/Dysgu
mkdir ${DIR}
cd ${DIR}
source ${CondaPATH}/bin/activate SV
while read ID || [[-n ${ID}]]
do
Bam = ${MissionPATH}/fastq/OUT/${ID}/$ID.realigner.bam
mkdir ${ID}
cd ${ID}
dysgu run -p $Thread $Ref ${DIR}/${ID}/tmp $Bam > ${DIR}/${ID}.Dysgu.vcf
cd ${DIR}
done < ${MissionPATH}/IndividualList.txt
7. SVcalling (Gridss):
Thread = ${TreadNumber}
Ref = ${RefferncePATH}/Sheep.fa
DIR = ${MissionPATH}/Gridss2
mkdir ${DIR}
cd ${DIR}
source ${CondaPATH}/bin/activate SV
GRDS = ${CondaPATH}/envs/SV/bin/gridss
while read ID || [[-n ${ID}]]
do
Bam = ${MissionPATH}/fastq/OUT/${ID}/$ID.realigner.bam
mkdir ${ID}
cd ${ID}
${GRDS} --reference $Ref --output ${DIR}/${ID}.Gridss2.vcf --threads $Thread --jar ${PGridss}/gridss-2.13.2-gridss-jar-with-dependencies.jar --workingdir ${DIR}/${ID}/tmp $Bam
cd ${DIR}
done < ${MissionPATH}/IndividualList.txt
8. SVcalling (manta):
Ref = ${RefferncePATH}/Sheep.fa
DIR = ${MissionPATH}/Manta
mkdir ${DIR}
cd ${DIR}
source ${CondaPATH}/bin/activate SV
while read ID || [[-n ${ID}]]
do
Bam = ${MissionPATH}/fastq/OUT/${ID}/$ID.realigner.bam
mkdir ${ID}
cd ${ID}
configManta.py --bam $Bam --referenceFasta $Ref --runDir ${DIR}/${ID}
python ${DIR}/${ID}/runWorkflow.py
gunzip ${DIR}/${ID}/results/variants/candidateSV.vcf.gz
cat ${DIR}/${ID}/results/variants/candidateSV.vcf > ${DIR}/${ID}.Manta.vcf
cd ${DIR}
done < ${MissionPATH}/IndividualList.txt
9. SVcalling (smoove):
Thread = ${TreadNumber}
Ref = ${RefferncePATH}/Sheep.fa
DIR = ${MissionPATH}/Smoove
mkdir ${DIR}
cd ${DIR}
source ${CondaPATH}/bin/activate SV
while read ID || [[-n ${ID}]]
do
Bam = ${MissionPATH}/fastq/OUT/${ID}/$ID.realigner.bam
mkdir ${ID}
cd ${ID}
smoove call --outdir ${DIR}/${ID}/ --name ${ID} --fasta $Ref -p $Thread --genotype $Bam
gunzip ${DIR}/${ID}/$ID\-smoove.genotyped.vcf.gz
cat ${DIR}/${ID}/$ID\-smoove.genotyped.vcf > ${DIR}/${ID}.Smoove.vcf
cd ${DIR}
done < ${MissionPATH}/IndividualList.txt
10. SVcalling (wham):
Thread = ${TreadNumber}
Ref = ${RefferncePATH}/Sheep.fa
DIR = ${MissionPATH}/Wham
mkdir ${DIR}
cd ${DIR}
source ${CondaPATH}/bin/activate SV
while read ID || [[-n ${ID}]]
do
Bam = ${MissionPATH}/fastq/OUT/${ID}/$ID.realigner.bam
mkdir ${ID}
cd ${ID}
whamg -x $Thread -a $Ref -f $Bam > Wham.vcf 2 > Wham.err
cat ${DIR}/${ID}/Wham.vcf > ${DIR}/${ID}.Wham.vcf
cd ${DIR}
done < ${MissionPATH}/IndividualList.txt
11. Candidate SV (SURVIVOR)
source ${CondaPATH}/bin/activate SV
Tools = “Delly Dysgu Gridss2 Manta Smoove Wham”
while read ID;
do
for TLS in $Tools
do
rm -rf ${MissionPATH}/SVmerge/TMP
mkdir ${MissionPATH}/SVmerge/TMP
cd ${MissionPATH}/SVmerge/TMP
cp ${MissionPATH}/$TLS/$ID.$TLS.vcf SV.vcf
find ${MissionPATH}/SVmerge/TMP/*.vcf > List.txt
SURVIVOR merge List.txt 50 1 1 1 0 50 OUT.vcf
grep -v “SVTYPE = TRA” OUT.vcf > OUT.noTRA.vcf
SURVIVOR filter OUT.noTRA.vcf NA 50 100000 0 -1 OUT.filter.vcf
grep -v “#“ OUT.filter.vcf | awk ‘{print $NF}’ | awk -F “:” ‘{print $7,$9,$10,$11}’ > OUT.Split1.txt
sed ‘s/,/ /g’ OUT.Split1.txt | awk ‘{for(i = 1; i < = (NF-4)/2 + 1; i ++ ){print $i,$(NF/2),$(1 + (NF/2)),$(i + (NF/2) + 1)}}’ > OUT.Split2.txt
sed ‘s/_/\t/g’ OUT.Split2.txt | sed ‘s/-/\t/g’ | awk ‘{if($5 < $7){print $4”:”$5”-“$7,$1,$2,$3}else{print $4”:”$7”-“$5,$1,$2,$3}}’ | sort | uniq > OUT.Split2.order.txt
awk ‘{print $1,ID,TLS,$2,$3,$4}’ ID = $ID TLS = $TLS OUT.Split2.order.txt | sed ‘s/ /\t/g’ » ${MissionPATH}/SVmerge/01-Merge/$ID.out
done
done < ${MissionPATH}/IndividualList.txt
cat ${MissionPATH}/SVmerge/01-Merge/*.out > ${MissionPATH}/SVmerge/01-Merge.txt
cut -f 1,4 01-Merge.txt | grep -v “NW:” | sort | uniq -c | sed ‘s/^ *//g’ | sed ‘s/ /\t/g’ | sort -nr -k1 > 01-Merge.sort.txt
awk ‘{if($1 > 3){print $2”\t”$3”\t”$1}}’ 01-Merge.sort.txt | sed ‘s/:/\t/g’ | sed ‘s/-/\t/g’ | sort -k1,1 -k2n,2 > 01-Merge.sort.bed
12. Candidate SV (cluster)
source ${CondaPATH}/bin/activate SV
cd ${MissionPATH}/SVmerge
grep “INS” 01-Merge.sort.bed | grep -v “NC” > 02-Merge.INS.bed
grep -v “INS” 01-Merge.sort.bed | grep -v “NC” > 02-Merge.nonINS.bed
bedtools intersect -a 02-Merge.INS.bed -b 02-Merge.INS.bed -wa -wb > 02-Merge.INS.link.txt
bedtools intersect -a 02-Merge.nonINS.bed -b 02-Merge.nonINS.bed -wa -wb > 02-Merge.nonINS.link.txt
awk ‘{print $1”:”$2”:”$3,$6”:”$7”:”$8}’ 02-Merge.INS.link.txt | awk ‘{if(M[$1]! = 1 & & M[$2] = = 1){A[$1] = A[$2];M[$1] = 1};if(M[$1] = = 1 & & M[$2]! = 1){A[$2] = A[$1];M[$2] = 1};if(M[$1]! = 1 & & M[$2]! = 1){S + = 1;ID = “TYPE“S;A[$1] = ID;A[$2] = ID;M[$1] = 1;M[$2] = 1};print $1,A[$1];print $2,A[$2]}’ | sed ‘s/ /\t/g’ | sort -k3,3 -k1n,1 -k2n,2 | uniq | sed ‘s/:/ /g’ > 02-Merge.INS.class.txt
awk ‘{print $1”:”$2”:”$3,$6”:”$7”:”$8}’ 02-Merge.nonINS.link.txt | awk ‘{if(M[$1]! = 1 & & M[$2] = = 1){A[$1] = A[$2];M[$1] = 1};if(M[$1] = = 1 & & M[$2]! = 1){A[$2] = A[$1];M[$2] = 1};if(M[$1]! = 1 & & M[$2]! = 1){S + = 1;ID = “TYPE“S;A[$1] = ID;A[$2] = ID;M[$1] = 1;M[$2] = 1};print $1,A[$1];print $2,A[$2]}’ | sed ‘s/ /\t/g’ | sort -k3,3 -k1n,1 -k2n,2 | uniq | sed ‘s/:/ /g’ > 02-Merge.nonINS.class.txt
awk ‘ARGIND = = 1{A[$1][$2][$3] = $4}ARGIND = = 2{print A[$1][$2][$3],$0}’ 02-Merge.INS.class.txt 02-Merge.INS.bed | sed ‘s/ /\t/g’ | sort -k1,1 -k6nr,6 > 02-Merge.INS.class.bed
awk ‘ARGIND = = 1{A[$1][$2][$3] = $4}ARGIND = = 2{print A[$1][$2][$3],$0}’ 02-Merge.nonINS.class.txt 02-Merge.nonINS.bed | sed ‘s/ /\t/g’ | sort -k1,1 -k6nr,6 > 02-Merge.nonINS.class.bed
13. Breakpoint (INS)
awk ‘{if($4 = = “INS” & & $5! = “NA” & & $6! = “Na”){print $1,$5,$6}}’ 01-Merge.txt | grep -v “NW:” | sort | uniq -c | sed ‘s/^ *//g’ | sed ‘s/ /\t/g’ | sort -k2,2 -k1nr,1 | awk ‘{if(M[$2]! = 1){print};M[$2] = 1}’ > 03-INS.sequence.txt
sed ‘s/:/\t/g’ 03-INS.sequence.txt | sed ‘s/-/\t/g’ | awk ‘ARGIND = = 1{A[$2][$3][$4] = $5”\t”$6}ARGIND = = 2{print $0”\t“A[$2][$3][$4]}’ - 02-Merge.INS.class.bed > 03-INS.annotation.txt
awk ‘{if($7! = “”){print}}’ 03-INS.annotation.txt | awk ‘{if(M[$1]! = 1){print};M[$1] = 1}’ | sort -k2n,2 -k3n,3 > 03-INS.candidates.txt
#Non INS
awk ‘ARGIND = = 1{A[$1][$5] + = $6}ARGIND = = 2{print $1”\t”$5”\t“A[$1][$5]}’ 02-Merge.nonINS.class.bed 02-Merge.nonINS.class.bed | sort | uniq | sort -k1,1 -k3nr,3 | awk ‘{if(M[$1]! = 1){print};M[$1] = 1}’ > 03-NonINS.class.txt
awk ‘ARGIND = = 1{L[$1] = $2}ARGIND = = 2{A[$1][$2] = 1}ARGIND = = 3{if(A[$1][$5] = = 1 & & $4 < L[$2] & & $3 > 2)print}’ 03-ref.fa.fai 03-NonINS.class.txt 02-Merge.nonINS.class.bed | awk ‘{if(M[$1]! = 1){print}M[$1] = 1}’ | awk ‘{if($6 > 3)print}’ > 03-NonINS.class.filter.txt
awk ‘{if($5 = = “DEL”)print $2”\t”$3”\t”$4}’ 03-NonINS.class.filter.txt | sort -k1n,1 -k2n,2 > 03-NonINS.class.filter.DEL.txt
awk ‘{if($5 = = “INV”)print $2”\t”$3”\t”$4}’ 03-NonINS.class.filter.txt | sort -k1n,1 -k2n,2 > 03-NonINS.class.filter.INV.txt
awk ‘{if($5 = = “DUP”)print $2”\t”$3”\t”$4}’ 03-NonINS.class.filter.txt | sort -k1n,1 -k2n,2 > 03-NonINS.class.filter.DUP.txt
14. Vcf construction (INS)
Ref = ${RefferncePATH}/Sheep.fa
Candidates = 03-INS.candidates.txt
awk ‘{print $2”\t”$3-2”\t”$3-1}’ $Candidates | grep -v “NN” | bedtools getfasta -fi $Ref -bed - | tr ‘a-z’ ‘A-Z’ | grep -v “ > ” | paste - $Candidates | awk ‘ OFS = “\t” {print $3,$4-1,“INS“NR,$1,$1$9,”.”,”.”,“DP = 1”,“GT:DP”,“1/1”}’ > 04-INS.vcf
15. Vcf construction (DEL)
Candidates = 03-NonINS.class.filter.DEL.txt
awk ‘{print $1,$2-2,$3}’ $Candidates | sed ‘s/ /\t/g’ | bedtools getfasta -fi $Ref -bed - | tr ‘a-z’ ‘A-Z’ | grep -v “ > “ > 04-DEL.ref.fa
awk ‘{print $1,$2-2,$2-1}’ $Candidates | sed ‘s/ /\t/g’ | bedtools getfasta -fi $Ref -bed - | tr ‘a-z’ ‘A-Z’ | grep -v “ > “ > 04-DEL.alt.fa
paste $Candidates 04-DEL.ref.fa 04-DEL.alt.fa | grep -v “NN” | awk ‘{print $1,$2-1,“DEL“NR,$4,$5,”.”,”.”,“DP = 1”,“GT:DP”,“1/1”}’ | sed ‘s/ /\t/g’ > 04-DEL.vcf
cat 04-INS.vcf 04-DEL.vcf | sort -k1,1 -k2n,2 > 05-Merge.vcf
16. Genotyping (vcf spilting)
mkdir ${MissionPATH}/GraphTyper
cd ${MissionPATH}/GraphTyper
cat ${MissionPATH}/SVmerge/05-Merge.vcf > ./SVpos.vcf
source ${CondaPATH}/bin/activate SV
while read CHR LEN
do
mkdir ${CHR}
cd ${CHR}
vcftools --vcf../SVpos.vcf --chr ${CHR} --recode --recode-INFO-all --stdout | bgzip -c > ./SV.${CHR}.vcf.gz
bcftools index -t SV.${CHR}.vcf.gz
cd ..
done < ${RefferncePATH}/Sheep.fa.fai
17. Genotyping (CHRgenotyping)
while read CHR LEN
do
DIR = ${MissionPATH}/GraphTyper/${CHR}
REF = ${RefferncePATH}/Sheep.fa
GTR = ${CondaPATH}/envs/SV/bin/graphtyper
VCF = SV.${CHR}.vcf.gz
BAMLIST = ${MissionPATH}/BamList.txt
REG = ${CHR}
THRD = 24
mkdir ${DIR}
cd ${DIR}
source ${CondaPATH}/bin/activate SV
${GTR} genotype_sv ${REF} ${VCF} --sams = ${BAMLIST} --region = ${REG} --threads = ${THRD} --log = Mission.log --vverbose
done < ${RefferncePATH}/Sheep.fa.fai
18. Merge
mkdir ${MissionPATH}/GraphTyper/chrAll
cd ${MissionPATH}/GraphTyper/chrAll
find ${MissionPATH}/GraphTyper/*/sv_results/*/*.vcf.gz | sort > pre.NC_All.txt
bcftools concat --file-list pre.NC_All.txt -Oz -o pre.NC_All.vcf.gz
zgrep “#“ pre.NC_All.vcf.gz > AGGREGATED.NC_All.vcf
zgrep -v “#“ pre.NC_All.vcf.gz | grep “AGGREGATED” >> AGGREGATED.NC_All.vcf
bgzip AGGREGATED.NC_All.vcf
bcftools index -t AGGREGATED.NC_All.vcf.gz
plink --vcf AGGREGATED.NC_All.vcf.gz --allow-extra-chr --keep-allele-order --recode vcf-iid --geno 0.3 --out g3.allSV --make-bed
bgzip g3.allSV.vcf
plink --vcf g3.allSV.vcf.gz --allow-extra-chr --keep-allele-order --missing --out g3.allSV
References
Li, X. et al. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat Commun. 11, 2815, https://doi.org/10.1038/s41467-020-16485-1 (2020).
Dos Santos, A. C. S. et al. A comparative study on the excretion of urinary metabolites in goats and sheep to evaluate spot sampling applied to protein nutrition trials. J Anim Sci. 96, 3381–3397, https://doi.org/10.1093/jas/sky198 (2018).
McHugh, N., Pabiou, T., McDermott, K., Wall, E. & Berry, D. P. A novel measure of ewe efficiency for breeding and benchmarking purposes. J Anim Sci. 96, 2051–2059, https://doi.org/10.1093/jas/sky143 (2018).
Godfray, H. C. J. et al. Meat consumption, health, and the environment. Science. 361, eaam5324, https://doi.org/10.1126/science.aam5324 (2018).
He, M. et al. Effects of YAP1 on proliferation and differentiation of Hu sheep skeletal muscle satellite cells in vitro. Anim Biotechnol. 34, 2691–2700, https://doi.org/10.1080/10495398.2022.2112688 (2023).
Chen, K. et al. Genomic insights into demographic history, structural variation landscape, and complex traits from 514 Hu sheep genomes. J Genet Genomics. 52, 245–257, https://doi.org/10.1016/j.jgg.2024.11.015 (2025).
Lin, C. et al. Expression and polymorphisms of CD8B gene and its associations with body weight and size traits in sheep. Anim Biotechnol. 34, 1214–1222, https://doi.org/10.1080/10495398.2021.2016432 (2023).
Weischenfeldt, J., Symmons, O., Spitz, F. & Korbel, J. O. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat Rev Genet. 14, 125–138, https://doi.org/10.1038/nrg3373 (2013).
Han, B. et al. Multiomics Analyses Provide New Insight into Genetic Variation of Reproductive Adaptability in Tibetan Sheep. Mol Biol Evol. 41, msae058, https://doi.org/10.1093/molbev/msae058 (2024).
Yang, J. et al. Structural variant landscapes reveal convergent signatures of evolution in sheep and goats. Genome Biol. 25, 148, https://doi.org/10.1186/s13059-024-03288-6 (2024).
Liang, X. Genomic structural variation contributes to evolved changes in gene expression in high-altitude Tibetan sheep. Proc Natl Acad Sci USA. 121, e2322291121, https://doi.org/10.1073/pnas.2322291121 (2024).
Al-Mamun, H. A. et al. Genome-wide association study of body weight in Australian Merino sheep reveals an orthologous region on OAR6 to human and bovine genomic regions affecting height and weight. Genet Sel Evol. 47, 66, https://doi.org/10.1186/s12711-015-0142-4 (2015).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Davenport, K. M. et al. An improved ovine reference genome assembly to facilitate in-depth functional annotation of the sheep genome. Gigascience. 11, giab096, https://doi.org/10.1093/gigascience/giab096 (2022).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997 (2013).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience. 10, giab008, https://doi.org/10.1093/gigascience/giab008 (2021).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303, https://doi.org/10.1101/gr.107524.110 (2010).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81, 559–575, https://doi.org/10.1086/519795 (2007).
Browning, B. L., Tian, X., Zhou, Y. & Browning, S. R. Fast two-stage phasing of large-scale sequence data. Am J Hum Genet. 108, 1880–1890, https://doi.org/10.1016/j.ajhg.2021.08.005 (2021).
Rausch, T. et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 28, i333–i339, https://doi.org/10.1093/bioinformatics/bts378 (2012).
Cleal, K. & Baird, D. M. Dysgu: efficient structural variant calling using short or long reads. Nucleic Acids Res. 50, e53, https://doi.org/10.1093/nar/gkac039 (2022).
Cameron, D. L. et al. GRIDSS2: comprehensive characterisation of somatic structural variation using single breakend variants and structural variant phasing. Genome Biol. 22, 202, https://doi.org/10.1186/s13059-021-02423-x (2021).
Chen, X. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 32, 1220–1222, https://doi.org/10.1093/bioinformatics/btv710 (2016).
Kronenberg, Z. N. et al. Wham: Identifying Structural Variants of Biological Consequence. PLoS Comput Biol. 11, e1004572, https://doi.org/10.1371/journal.pcbi.1004572 (2015).
Pedersen, B. S., Layer, R. & Quinlan, A. R. smoove: Structural-variant calling and genotyping with existing tools. Figshare https://github.com/brentp/smoove (2020).
Eggertsson, H. P. et al. Graphtyper enables population-scale genotyping using pangenome graphs. Nat Genet. 49, 1654–1660, https://doi.org/10.1038/ng.3964 (2017).
Yin, L. et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Genomics Proteomics Bioinformatics. 19, 619–628, https://doi.org/10.1016/j.gpb.2020.10.007 (2021).
Hu, Z. L., Park, C. A. & Reecy, J. M. Bringing the Animal QTLdb and CorrDB into the future: meeting new challenges and providing updated services. Nucleic Acids Res. 50, D956–D961, https://doi.org/10.1093/nar/gkab1116 (2022).
Fonseca, P. A. S., Suárez-Vega, A., Marras, G. & Cánovas, Á. GALLO: An R package for genomic annotation and integration of multiple data sources in livestock for positional candidate loci. Gigascience. 9, giaa149, https://doi.org/10.1093/gigascience/giaa149 (2020).
Ding, L. et al. The HTIRDB: A resource containing a transcriptional atlas for 105 different tissues from each of seven species of domestic herbivore. Imeta. 4, e267, https://doi.org/10.1002/imt2.267 (2025).
Szpiech, Z. A. & Hernandez, R. D. selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol Biol Evol. 31, 2824–2827, https://doi.org/10.1093/molbev/msu211 (2014).
Biscarini, F., Cozzi, P., Gaspa, G. & Marras, G. detectRUNS: an R package to detect runs of homozygosity and heterozygosity in diploid genomes. Figshare https://cran.r-project.org/web//packages//detectRUNS/vignettes/detectRUNS.vignette.html (2019).
NGDC BioProject database https://ngdc.cncb.ac.cn/gsa/browse/CRA017832 (2025).
European Variation Archive https://www.ebi.ac.uk/eva/?eva-study=PRJEB94328 (2025).
Acknowledgements
This research was supported by grants from Biological Breeding-National Science and Technology Major Project (2022ZD04017), Zhejiang Province Agricultural Major Technology Collaborative Promotion Project (2025ZDXT15), the Zhejiang Science and Technology Major Program on Agricultural New Variety Breeding (2021C02068-6), National Natural Science Foundation of China (32172724), and Zhejiang Team Technology Ambassador Project (Tongxiang).
Author information
Authors and Affiliations
Contributions
Z.G.W. conceived and designed the experiments. X.X. designed the analytical strategy and wrote the manuscript. Y.J.G. submitted the dataset. L.R.Z assisted in writing the manuscript. W.G. and M.L. collected and prepared sequencing samples. P.J.Z. revised the paper. All authors have read and agreed to submit the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiang, X., Guo, Y., Zhao, L. et al. Whole genome sequencing and structural variations provide insights into the body size traits of Hu sheep. Sci Data 12, 1373 (2025). https://doi.org/10.1038/s41597-025-05734-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05734-x
