
Abstract
As a protandrous hermaphroditic fish species with natural sex change from male to female, Asian seabass (Lates calcarifer) represents an attractive model for studying sequential hermaphroditism. In this study, we constructed the first telomere-to-telomere (T2T) gap-free genome assembly of Asian seabass, by integration of MGI short-read, PacBio HiFi long-read, ONT ultra-long and Hi-C sequencing technologies. The haplotypic 614.19 Mb genome sequences were successfully anchored onto 24 chromosomes, demonstrating exceptional contiguity with a contig N50 of 26.57 Mb. Comprehensive annotation revealed precise localization of telomeric repeats and centromeric regions across various chromosomes. Good results from Merqury (QV: 57.8), CRAQ (99.45%) and BUSCO (100%) indicate a high level of accuracy for the assembled genome. ONT ultra-long and PacBio HiFi sequencing data were aligned with the assembly using minimap2, resulting in a mapping rate over 98%. Repetitive elements accounted for 18.18% (111.64 Mb) of the entire genome, and a total of 25,093 protein-coding genes were annotated. This high-quality T2T genome assembly provides a valuable genetic resource for in-depth comparative genomics, population genetics, molecular breeding, and functional studies of this economically important marine species. This reference assembly also facilitates investigations into the detailed molecular mechanisms underlying its unique reproductive strategy of the protandrous hermaphrodite Asian seabass.
Similar content being viewed by others

Background & Summary
Sex determination is a genetic or epigenetic process that initiates and regulates the developmental trajectory of sexual differentiation, whereas sex differentiation encompasses the cascade of morphological and physiological events through which a bi-potential gonad progressively develops into either a testis or an ovary, culminating in the establishment of species-specific secondary sexual characteristics1. Compared with those highly conserved sex determination systems in various mammals and birds, fishes exhibit remarkable diversity in sex determination patterns. They present more diversified sex determination modes than higher vertebrates, such as genetic sex determination (GSD), environmental sex determination (ESD), and the coexistence of both2,3. Notably, among diverse environmental cues, temperature emerges as the most influential exogenous factor to modulate sexual development in fishes. Numerous species across different taxa have been documented to own thermally sensitive sex determination, where incubation temperature during critical developmental windows can override genotypic sex determinants. Good examples include European seabass (Dicentrarchus labrax)4, tilapia (Nile tilapia and Oreochromis niloticus)5, and Atlantic halibut (Hippoglossus hippoglossus)6,7. These fishes exhibit interesting characteristics of temperature-dependent sex determination, and their sex ratios can change significantly with variations in environmental temperature during their hatching period.
In addition to gonochorism (separate sexes), fishes also exhibit hermaphroditism as an important reproductive strategy. Approximately 2% of teleost fishes are hermaphroditic, distributed across 27 families within 7 orders8. Sex change is a biological process in which an organism transitions from its original sex to another through specific physiological mechanisms. Organisms capable of naturally undergoing sex change are referred to hermaphrodites, which are typically categorized into protandrous (male-to-female) and protogynous (female-to-male)9. Common examples in these fishes include groupers, black seabream, clownfish, and ricefield eel10,11,12,13.
Asian seabass holds substantial cultural and economic values throughout the tropical Indo-West Pacific region, serving as both a key fishery resource and a commercially important aquaculture species14. As a protandrous hermaphroditic fish15, it usually first develops into a male at 3–4 years of age, and then approximately 90% of individuals undergoes natural sex change to female by age 616. Despite its remarkable reproductive strategy, the genetic mechanisms underlying sex change in Asian seabass remain poorly understood, as is the case for most hermaphroditic species. Genomic resources, including DNA markers, high-resolution linkage maps, transcriptomes, reference genome sequences along with their comprehensive annotations, play a pivotal role in supporting aquaculture. These valuable genetic resources provide a solid foundation for diverse applications, enabling comprehensive genetic investigations to support development of sophisticated artificial breeding strategies. Ultimately, they contribute to the sustainable expansion and increased productivity of international aquaculture industry14. Given the economic value of Asian seabass and its remarkable natural sex change, construction of its high-quality genome assembly is absolutely essential.
In this study, we combined MGI short-read, PacBio HiFi long-read, ONT (Oxford Nanopore Technologies) ultra-long, and Hi-C sequencing data to generate a high-fidelity T2T genome assembly of Asian seabass. This assembly was rigorously assessed for quality, and its key genomic features were systematically characterized. In fact, this gap-free and complete reference assembly represents a substantial improvement over any previous assembly of this species17. It will not only facilitate population genetic research and evolutionary study, but also provide an important genetic resource for molecular breeding and investigating molecular mechanisms of sex change in this economically important fish.
Methods
Sample collection
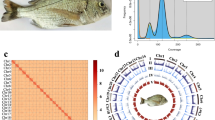
A male Asian seabass (Fig. 1A) was collected from a local aquaculture facility of the South China Sea Fisheries Research Institute under Chinese Academy of Fishery Sciences, which is located in Guangzhou City, Guangdong Province, China. Muscle tissue was sampled for whole-genome sequencing, including MGI short read, PacBio HiFi long read, ONT (Oxford Nanopore Technologies) Ultra-long and Hi-C sequencing technologies. Additionally, seven distinct tissues (such as gill, brain, liver, muscle, eye, testis, and skin) were collected for transcriptome sequencing (Table 1). Upon dissection into small fragments, the tissue samples were washed with ice-cold PBS (pH 7.4) to eliminate blood residues and contaminants. After removing outside liquid by blotting, these samples were rapidly frozen in liquid nitrogen and subsequently maintained at −80 °C before use. For transcriptome sequencing, frozen specimens were shipped in dry ice containers to the sequencing company (BGI, Shenzhen, Guangdong, China).

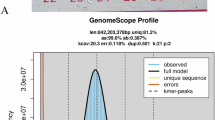
Asian seabass and its whole-genome sequence distribution. (A) A morphological image of the sequenced Asian seabass. (B) A k-mer (21-mer) distribution curve for estimation of the genome size.
DNA extraction and genome sequencing
Genomic DNA (gDNA) was extracted from muscle tissue using a QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocols18. Fragment size, purity, and quantification of the extracted gDNA were assessed via 0.75% agarose gel electrophoresis, an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) and a Qubit Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA), respectively.
For the MGI short-read sequencing, gDNA was randomly fragmented using a MGIEasy Universal DNA Library Preparation Kit (MGI, Shenzhen, China) to construct a library with an insert-size of 350 bp. Sequencing was performed on a DNBSEQ-T7 platform (MGI), generating 37.4 Gb of raw 150-bp paired-end reads, and then filtered by fastp v0.12.619 (parameter: -n 0 -f 5 -F 5 -t 5 -T 5) to remove adaptor sequences and low-quality reads. Finally, a total of 33.69 Gb of clean reads (Table 1) were obtained for further data error correction and genome-size estimation.
For the PacBio HiFi sequencing, approximately 10 μg of high-quality gDNA was applied to construct a SMRTbell library following the manufacturer’s standard protocol (SMRTbell Express Template Prep Kit 2.0; Pacific Biosciences, Menlo Park, CA, USA), which was then sequenced on a PacBio Sequel II System using the circular consensus sequencing (CCS) technology. A total of 90.47 Gb of HiFi reads with a N50 of 18,366 bp were obtained (Table 1) using the CCS v6.0.020 (Circular Consensus Sequencing) software with the optimized parameter “-min-passes 3”.
Two ultra-long read libraries were constructed using Oxford Nanopore Technologies (ONT) protocols, which were sequenced on a PromethION platform (Oxford Nanopore Technologies Co., Littlemore, Oxford, UK). Raw reads were initially processed to eliminate those with a quality value (QV) lower than 7 using the NanoFilt v2.8.021 software. Finally, a total of 1.54 million clean reads were retained, accumulating a substantial base count of 61.32 Gb. The average read length was 39.69 kb, with an N50 length of 71.17 kb (Table 1).
For the high-throughput chromosome conformation capture (Hi-C) sequencing, one Hi-C library was generated using a GrandOmics Hi-C kit (GrandOmics, Wuhan, Hubei, China) following the manufacturer’s protocol. In brief, gDNA was first cross-linked using a 4% formaldehyde solution to stabilize chromatin structures. Subsequently, the DNA was digested with the restriction enzyme MboI to introduce specific cleavage sites. Those resulting DNA fragments were then labeled with biotin-14-dCTP, allowing for incorporation of a detectable marker. The labeled DNA fragments were ligated using T4 DNA ligase to facilitate subsequent enrichment steps. Following ligation, the DNA was further digested to yield fragments in the size range of 200 to 600 bp. The library was sequenced on a DNBSEQ-T7 platform (MGI, Shenzhen, China) using a 150-bp paired-end model. The Hi-C sequencing technology generated 102.32 Gb of raw data. Subsequently, fastp v0.12.619 was applied to filter adaptor sequences and low-quality reads. Finally, 93.8 Gb of Hi-C clean data were retained (Table 1) for chromosome assembly.
RNA extraction and transcriptome sequencing (RNA-seq)
Total RNA was extracted from seven tissues separately according to a standard Trizol protocol (Invitrogen, Frederick, MD, USA), followed by purification with a Qiagen RNeasy Mini Kit (Qiagen, Germantown, MD, USA). RNA concentration and integrity were measured using a NanoDrop 8000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), respectively. Only those RNA samples with OD260/280 ≥ 1.8 and RNA integrity ≥ 7.0 were selected for transcriptome sequencing. RNA was used for construction of a cDNA library followed the manufacture’s guideline, which was then sequenced on a HiSeq X Ten platform (Illumina, San Diego, CA, USA). A total of 48.07 Gb of transcriptome raw data were generated (Table 1), which aided in annotation of protein-coding genes and prediction of gene structures.
Genome-size estimation and construction of a T2T genome assembly
To estimate the genome size of Asian seabass, we employed jellyfish (v2.2.10)22 to perform k-mer counting with k = 21, and the parameters were set as ‘-m 21 -s 10 G -C’. Subsequently, a generated histogram was utilized as an input file for GenomeScope v2.023 to estimate genetic characteristics. This approach provided a sequence-derived estimate of the Asian seabass genome characteristics prior to assembly. Our analysis results show that the genome size of Asian seabass is approximately 576.74 Mb, with an estimated heterozygosity of about 0.46% (Fig. 1B) and repetitive sequences accounting for 32.79 Mb (5.69%).
Primary contigs were initially generated by assembling PacBio HiFi and ONT data using Hifiasm v0.19.824 with default parameters. Then, purge_dups v1.2.525 was employed to remove haplotypic and heterozygous duplications from the de novo assembly, yielding a final assembly with a total length of 614.08 Mb.
Using the preliminary assembly as the reference, Hi-C clean reads were utilized to construct chromosomes for Asian seabass. First, the Hi-C reads were mapped to the assembled contigs using bowtie2 v2.2.5 (–very-sensitive -L 20–score-min L, −0.6, −0.2–end-to-end)26. Subsequently, the HiC-Pro v2.8.127 pipeline was applied to detect ligation products, retaining only valid paired reads for downstream analysis. Based on these valid reads, the primary assembly was clustered, ordered, and oriented into chromosomes using the Juicer v1.528 and 3D-DNA v3.029 software with parameters -m haploid -r 2 -c 24. Juicebox v1.11.0830 was employed to visualize before manually adjusting the candidate assemblies.
To fill the remaining gaps, those corrected ultra-long ONT reads were applied to generate a gap-free genome assembly using TGS-GapCloser v1.2.131 with optimized parameter “–min_match 1000–min_nread 3” and LR_Gapcloser v1.032 with the parameter “-t 35 -m 1000000 -v 500”. The final genome assembly spans 614.19 Mb, and it is anchored onto 24 chromosomes (Fig. 2), among them the longest and the shortest are 31.85 Mb and 14.85 Mb, respectively (Table 2).

The first T2T genome assembly of Asian seabass. (A) Genome-wide chromatin interactions at a 500-kb resolution. Color blocks represent corresponding interactions, with various strengths from yellow (low) to red (high). (B) A Circos plot of the main genome features. From outside to inside include the 24 chromosomes, gene density, GC content, repetitive sequences density, and a colinear relationship among chromosomes of the Asian seabass genome assembly. Note that the density calculation window is set as 100 kb.
Identification of the centromere and telomere sequences
Telomeres were identified by searching for the target sequence (CCCTAA/TTAGGG) at both ends of each chromosome using Telomere-to-Telomere Toolkit quarTeT v1.1.133. Centromeres, as specialized DNA sequences connecting sister chromatids, exhibit complex structures in most animals and plants with highly repetitive satellite DNA and scattered retrotransposon sequences. In this study, after identifying repeat sequences according to TRF v4.0.434 and RepeatMasker v4.0.635 and obtaining a TE annotation file, quarTeT v1.1.133 was applied to identify centromeres, and the candidate interval range of every centromere was predicted. Ultimately, we determined that the Asian seabass genome contains a complete set of 24 centromeres and 48 telomeres (Table 3; Fig. 3).

Genome-wide localization of repetitive elements (REs), telomeres and centromeres. The triangles at both ends of each chromosome represent the telomere regions, and the gully area within each chromosome stands for the centromere region.
Annotation of repeat elements
For prediction of repetitive elements (REs), tandem repeats were first annotated using TRF v4.0.434 and GMATA v2.236. TRF was employed to identify simple sequence repeats (SSRs), whereas GMATA was used to recognize all tandem REs across the entire genome.
Transposable elements (TEs) in the assembled genome were predicted using a combination of homology-based and de novo methods. For the homology approach, TEs were identified using RepeatMasker v4.0.6 and RepeatProteinMask v4.0.635. For the de novo approach, RepeatModeler v1.0.837 and LTR_FINDER v1.0.638 were employed to generate a de novo repeat library, and RepeatMasker was applied to annotate REs against this repeat library. The annotation results of all repetitive sequences were merged into a comprehensive dataset. This comprehensive annotation revealed 111.64 Mb of repetitive sequences, which account for 18.18% of the assembled Asian seabass genome (Fig. 3). The most abundant repetitive element was DNA transposons at 9.00% (55.26 Mb), followed by long interspersed nuclear elements (LINEs) at 2.89% (17.76 Mb) and long terminal repeats (LTRs) at 2.46% (15.07 Mb) (see Table 4).
Prediction and functional annotation of protein-coding genes
Repetitive regions of the assembled genome were masked prior to prediction of genes and their structures. Protein-coding genes was annotated by combination of three methods, including de novo, homology and RNA-seq-based annotations. First, AUGUSTUS v3.2.139 and GlimmerHMM v3.0.440 were employed to perform the ab inito gene structure prediction. Second, GeMoMa v1.6.441 was applied for the homology-based prediction. We aligned homology proteins from five representative fish species, including Epinephelus fuscoguttatus (brown-marbled grouper, GCA_011397635.1), Epinephelus moara (kelp grouper, GCA_006386435.1), Lates japonicus (Japanese lates, GCA_033238685.1), Perca flavescens (yellow Perch, GCA_004354835.1) and Sebastes umbrosus (Honeycomb rockfish, GCA_015220745.1) downloaded from the NCBI. Third, the RNA-seq data from seven tissues were assembled into contigs using Trinity v2.5.142, and then gene structures were identified using PASA v2.3.343. Finally, gene sets were integrated by the Evidence Modeler (EVM) pipeline v1.044.
A total of 25,093 protein-coding genes were annotated, with an average gene length of 13.81 kb and an average coding sequence (CDS) length of 1,721.49 bp (Table 5). Protein-coding genes were evaluated using BUSCO with the actinopterygii_odb10 database as the reference. More than 98.8% of complete BUSCOs were identified within the predicted protein-coding genes.
Functional annotation of the protein-coding genes was performed using Blastp v2.2.2645, which aligned deduced protein sequences against five public databases including NCBI Non-Redundant Protein Sequence (NR), SwissProt46, Gene Ontology (GO)47, Kyoto Encyclopedia of Genes and Genomes (KEGG)48 and EuKaryotic Orthologous Groups (KOG)49, with an E-value cutoff of <1e−5. Ultimately, 23,711 protein-coding genes (94.49% of the total predicted genes) were functionally annotated, with at least one hit for each gene in the searched databases (Table 6).
Data Records
Files of the MGI, PacBio, ONT, Hi-C and transcriptome sequencing, and the assembled genome for Asian seabass were deposited at NCBI under the accession number PRJNA1245135. Raw reads are available in the Sequence Reads Archive (SRA) with the accession numbers SRR32997291 to SRR3299730550. The genome assembly, predicted coding sequences and function annotation files of Asian seabass were stored in Figshare (No: m9.figshare.28735226)51. The genome assembly has also been deposited at the NCBl/GenBank under the accession number of GCA_051027255.152.
Technical Validation
To evaluate the quality of our genome assembly, we employed four approaches. First, BUSCO v5.2.253 was employed to examine completeness. A total of 100% (single copy complete genes (S): 99.84%, duplicated complete genes (D): 0.16%) of complete BUSCOs in the actinopterygii_odb10 database were identified. Second, Merqury v1.32854 was applied to estimate the base-level accuracy and completeness on the basis of k-mer counts (generated from Illumina and PacBio HiFi reads), resulting in a QV of 40.59 and 57.80 respectively. Third, Clipping information for Revealing Assembly Quality (CRAQ, v1.09)55 was used to assess the accuracy of our genome assembly based on PacBio HiFi and Illumina reads, resulting in a R-AQI (assembly quality indicator) of 98.42 and a S-AQI of 99.45. Fourth, we mapped the sequencing data to the assembled genome using bwa v0.7.1756 and minimap2 v2.2657, which showed mapping rates of 99.46% for the MGI data, 99.99% for the PacBio data, and 98.43% for the ONT data. These results collectively support high quality of the Asian seabass genome assembly. The BUSCO completeness value was calculated to be 98.8% for the predicted protein-coding genes of Asian seabass (Table 7). To further evaluate the quality of these predicted protein-coding genes, we aligned the transcriptome data to the assembled genome using STAR v 2.7.11b58, and then calculated the exonic coverage rate with bedtools v2.29.259. We observed that 94.71% of the exonic regions had been covered with sequencing reads, indicating high annotation accuracy (see Table 7).
Code availability
The versions and parameters of bioinformatics tools applied in this study have been described in the Method section. If no parameter is provided, the default is set. No custom code was used.
References
Gamble, T. et al. Sex determination. Current Biology 22(8), 257–262 (2012).
Penman, D. J. et al. Fish gonadogenesis. Part I: genetic and environmental mechanisms of sex determination. Reviews in Fisheries Science 16(sup1), 16–34 (2008).
Devlin, R. H. et al. Sex determination and sex differentiation in fish: an overview of genetic, physiological, and environmental influences. Aquaculture 208(3-4), 191–364 (2002).
Piferrer, F. et al. Genetic, endocrine, and environmental components of sex determination and differentiation in the European sea bass (Dicentrarchus labrax L.). General and comparative endocrinology 142(1-2), 102–110 (2005).
Baroiller, J. F. et al. Tilapia sex determination: where temperature and genetics meet. Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology 153(1), 30–38 (2009).
Palaiokostas, C. et al. Mapping the sex determination locus in the Atlantic halibut (Hippoglossus hippoglossus) using RAD sequencing. BMC genomics 14, 1–12 (2013).
Hughes, V. et al. Effect of rearing temperature on sex ratio in juvenile Atlantic halibut, Hippoglossus hippoglossus. Environmental biology of fishes 81, 415–419 (2008).
Avise, J. C. et al. Evolutionary perspectives on hermaphroditism in fishes. Sexual Development 3(2-3), 152–163 (2009).
Kuwamura, T. et al. Sex change of primary males in a diandric labrid Halichoeres trimaculatus: coexistence of protandry and protogyny within a species. Journal of Fish Biology 70(6), 1898–1906 (2007).
Li, S. et al. Mechanisms of sex differentiation and sex reversal in hermaphrodite fish as revealed by the Epinephelus coioides genome. Molecular Ecology Resources 23(4), 920–932 (2023).
Zhang, K. et al. A telomere-to-telomere genome assembly of the protandrous hermaphrodite blackhead seabream, Acanthopagrus schlegelii. Scientific Data 12(1), 350 (2025).
Casas, L. et al. Sex change in clownfish: molecular insights from transcriptome analysis. Scientific Reports 6(1), 35461 (2016).
Cheng, H. et al. The rice field eel as a model system for vertebrate sexual development. Cytogenetic and Genome Research 101(3-4), 274–277 (2003).
Yue, G. H. et al. Genomic resources and their applications in aquaculture of Asian seabass (Lates calcarifer). Reviews in Aquaculture 15(2), 853–871 (2023).
Athauda, S. et al. Effect of rearing water temperature on protandrous sex inversion in cultured Asian Seabass (Lates calcarifer). General and Comparative Endocrinology 175(3), 416–423 (2012).
Jerry, D. R. Biology and culture of Asian seabass Lates calcarifer. CRC Press (2013).
Vij, S. et al. Chromosomal-level assembly of the Asian seabass genome using long sequence reads and multi-layered scaffolding. PLoS Genetics 12(4), e1005954 (2016).
Mei, L. et al. Evaluation of QIAamp® DNA Stool Mini Kit for ecological studies of gut microbiota. Journal of Microbiological Methods 54(1), 13–20 (2003).
Chen S. et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17), i884–i890.
Rhoads, A. et al. PacBio Sequencing and Its Applications. Genomics Proteomics & Bioinformatics 13, 278–289 (2015).
De Coster, W. et al. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34(15), 2666–2669 (2018).
Marçais, G. et al. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27(6), 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33(14), 2202–2204 (2017).
Cheng, H. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175 (2021).
Roach, M. J. et al. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19, 1–10 (2018).
Langmead, B. et al. Fast gapped-read alignment with Bowtie 2. Nature Methods 9(4), 357–359 (2012).
Dekker, J. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology 16, 259 (2015).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Systems 3(1), 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Systems 3, 99–101 (2016).
Xu, M. et al. TGS-GapCloser: a fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience 9(9), giaa094 (2020).
Xu, G. C. et al. LR_Gapcloser: a tiling path-based gap closer that uses long reads to complete genome assembly. Gigascience 8(1), giy157 (2019).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Horticulture Research 10(8), uhad127 (2023).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27(2), 573–580 (1999).
Tarailo-Graovac, M. et al. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics Chapter 4, 4–10 (2009).
Wang, X. & Wang, L. GMATA: an integrated software package for genome-scale SSR mining, marker development and viewing. Frontiers in Plant Science 7, 1350 (2016).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Science of the United States of America 117, 9451–9457 (2020).
Xu, Z. et al. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–268 (2007).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research 34, W435–439 (2006).
Majoros, W. H. et al. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20(16), 2878–2879 (2004).
Keilwagen, J. et al. GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods in Molecular Biology 1962, 161–177 (2019).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research 31(19), 5654–5666 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology 9, R7 (2008).
Altschul, S. F. et al. Basic local alignment search tool. Journal of Molecular Biology 215(3), 403–410 (1990).
Bairoch, A. et al. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Research 28(1), 45–48 (2000).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nature genetics 25(1), 25–29 (2000).
Kanehisa, M. et al. KEGG as a reference resource for gene and protein annotation. Nucleic acids research 44(D1), D457–D462 (2016).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP576768 (2025).
Zhang, X. Genome assembly, predicted coding sequences and functional annotation files of L. calcarifer. Figshare. https://doi.org/10.6084/m9.figshare.28735226 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_051027255.1 (2025).
Simao, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Rhie, A. et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21(1), 245 (2020).
Li, K. et al. Identification of errors in draft genome assemblies at single-nucleotide resolution for quality assessment and improvement. Nature Communications 14(1), 6556 (2023).
Li, H. et al. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14), 1754–1760 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34(18), 3094–3100 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1), 15–21 (2013).
Quinlan, A. R. et al. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6), 841–842 (2010).
Acknowledgements
This work was supported by Shenzhen Natural Science Foundation (no. JCYJ20241202124511016) and National Key Research and Development Program of China (no. 2022YFE0139700).
Author information
Authors and Affiliations
Contributions
Q.S. conceived and designed the study. X.Z., J.W. and J.C. collected the samples. X.Z., J.C. and J.W. performed data analysis. J.W. and W.Z. conducted experiments for species identification. X.Z. and J.W. wrote the manuscript. Q.S. revised the manuscript. All authors read and approved the final manuscript for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, X., Chen, J., Zhou, W. et al. A telomere-to-telomere gap-free genome assembly of the protandrous hermaphrodite Asian seabass (Lates calcarifer). Sci Data 12, 1457 (2025). https://doi.org/10.1038/s41597-025-05735-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05735-w
