
Abstract
Drosophila suzukii, an invasive pest native to East Asia, has spread globally over the past two decades, posing severe threats to soft-skinned fruits due to its strong reproductive capabilities and high adaptation. However, the absence of high-quality genomic resources has hindered a comprehensive understanding of its ecological adaptations and invasion strategies. Here, we present a chromosome-level genome assembly of D. suzukii using PacBio HiFi, Illumina short, and Hi-C data. The assembled genome size totals 157.35 Mb with a scaffold N50 of 25.66 Mb by anchoring 86.59% (136.24 Mb) of the assembly to six chromosomes. BUSCO analysis indicates completeness, with a score of 98.1% (n = 1,367), comprising 97.4% single-copy BUSCOs and 0.7% duplicated BUSCOs. The genome contains 17.41% (27.39 Mb) repetitive elements and encodes 14,742 predicted protein-coding genes. Comparative genomic analyses reveal strong chromosomal synteny between D. suzukii and Drosophila melanogaster. This high-quality genome assembly is a critical resource for elucidating the evolutionary dynamics, ecological adaptations, and invasive biology of D. suzukii, providing valuable insights into pest control strategies.
Similar content being viewed by others

Background & Summary
The spotted wing drosophila (Drosophila suzukii Matsumura) is an invasive pest characterized by rapid reproduction and a wide dispersal range1,2. Its distinctive serrated ovipositor is both large and robust, enabling it to pierce the skin of ripening and ripe soft-skinned fruits to lay eggs internally3,4. The larval development occurs entirely within the fruit, rendering infestation difficult to detect as no external signs are visible. Feeding by the larvae causes internal damage, often leading to secondary bacterial infections, accelerating fruit decay and severely compromising quality, yield, and market value2,5. Due to its high invasiveness, D. suzukii has emerged as a significant pest with devastating consequences for global fruit production, infesting the ripe fruits of nearly 200 plant species, including economically critical stone fruits and berries6.
D. suzukii was first described in a strawberry field in Japan in 19167, in China in 19378, and was later detected in Hawaii in 19809. Early reports indicated that D. suzukii did not cause significant economic losses and thus did not attract much attention at the time. However, D. suzukii was first reported in Europe and North America in 200810,11. And by the following year, it had caused substantial damage, drawing significant concern. D. suzukii has been detected across the stone fruit-growing regions, spanning from Southern California to British Columbia. It is estimated to cause a 50% yield loss in California raspberries, reducing total revenue by 37%, while processed strawberries could see a 20% drop in total income12. In 2009, it was also reported in several Mediterranean countries, including Spain, France, and Italy13, and it rapidly spread across multiple European countries in subsequent years. In 2010, D. suzukii led to losses of up to 80% in strawberry crops in certain areas of southern France. In Italy, particularly in blueberries, blackberries, and raspberries, losses ranged from 30% to 40%14. Since 2012, the pest has spread widely in South America and Africa13,14,15,16,17. It was first detected in Brazil and Uruguay in 2013 and in Argentina in 201415,18,19. In 2017, it was also reported in Morocco, where approximately 15% of small berries were infested20. Countries in Oceania, such as French Polynesia, have also reported infestations21. Currently, D. suzukii is reported as an invasive pest in over 50 countries worldwide21. Based on the environmental suitability models for D. suzukii, it is predicted that the species may further invade several regions, including Australia22.
D. suzukii has strong fecundity, a wide host range, and high dispersal potential, resulting in significant economic losses in fruit production1,11. High-quality genomic resources are crucial for advancing research to develop novel pest control strategies. A chromosome-level genome sequence of D. suzukii from Japan (NCBI GenBank assembly: GCF_043229965.1) was recently released. However, the analysis methods and data descriptions have not yet be fully detailed in a publication. Therefore, considering the ecological niche, geographic distribution, and invasion strategy of D. suzukii, it is important to resequence and analyze the genome with clearer methodologies.
Here, chromosome-level genome of D. suzukii was constructed by integrating PacBio HiFi, Illumina, and Hi-C datas. Non-coding RNAs, protein-coding genes, and repetitive elements were annotated. Furthermore, we investigated interspecific chromosomal variation between D. suzukii and D. melanogaster. The high-quality genome of D. suzukii serves as a valuable resource for advancing study of the evolution and ecological roles of Drosophila.
Methods
Samples preparation and sequencing
D. suzukii adults were collected in Jun 2018, from a cherry orchard in Yantai (37.41°N, 121.82°E), Shandong Province, China. Male D. suzukii were collected after PBS washing to avoid microbial contamination of body surfaces. The samples were conducted libraries and sequenced at Berry Genomics. Genomic DNA was extracted following CTAB method23. PacBio HiFi libraries with 15 kb insert size were constructed using the SMRTbeⅡ® Express Template Prep Kit 2.0 (Pacific Biosciences, California, USA) for sequencing on BacBio Sequel Ⅱ platform. For Illumina sequencing, Libraries with 350 bp insert size were prepared using the TruSeq DNA PCR-free kit for sequencing on the Illumina NovaSeq 6000 platform. Total RNA was extracted using the TRIzol™ Reagent, and RNA library was performed using the TruSeq RNA v2 kit. The Hi-C library was constructed by Berry Genomics Corporation. We collected a total of 78.56 Gb of sequencing data, comprising 37.22 Gb (236.54×) of Illumina reads, 12.28 Gb (70.05×) of PacBio HiFi reads, 16.31 Gb (103.67×) of Hi-C data, and 12.75 Gb of transcriptome data (Table 1). The PacBio HiFi reads had a scaffold N50 of 7.85 kb and an average length of 7.74 kb (Table S1).
Genome assembly
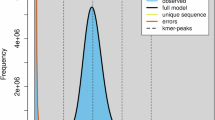
Initially, the Illumina sequencing data was processed for quality control using BBTools v38.8224. This process utilized two scripts, “clumpify.sh” and “bbduk.sh,” which were used to remove duplicate reads and perform quality control, including removing bases with quality scores below 20, filtering sequences shorter than 15 bp, and trimming poly-A/G/C tails longer than 10 bp. Subsequently, we conducted a genome K-mer analysis using GenomeScope v2.025 with “-k 21 -p 2 -m 1000”. The predicted genome size was estimated at 157.83 Mb, with a heterozygosity rate of 2.10% and repetitive sequences of 16.08 Mb (10.19%). The red peaks indicated potential contamination within the data (Fig. 1).

Genome survey at 21-mer of Drosophila suzukii estimated by GenomeScope. The vertical dotted lines represent the peaks of different coverages for heterozygous, homozygous, and the duplicated sequences separately.
The initial assembly of PacBio HiFi reads was performed using Hifiasm v0.16.126 with the parameter “-l 2”, which employs an “aggressive” method to remove redundant sequences. To minimize contamination and errors, contigs with sequencing depths exceeding 10 × were retained. To further improve assembly accuracy, Purge_Dups v1.2.527 was employed to remove heterozygous sequences, using BAM files generated by Minimap2 v2.2428 for alignment. After quality control with Juicer v1.6.229, Hi-C reads were assembled using the default parameters of 3D-DNA v18092230, followed by manual correction of the assembly with Juicebox v1.11.0829. Similarity searches were performed using MMseqs2 v1331 to identify potential contaminants. These searches targeted the NCBI nucleotide and UniVec databases with a sequence identity threshold of 0.8.
We obtained chromosome-level genome assembly of D. suzukii, which is 157.35 Mb in size and comprising 9 scaffolds and 209 contigs. The longest scaffold measured 33.65 Mb, while the longest contig was 7.75 Mb. The N50 values were 25.66 Mb for scaffolds and 1.74 Mb for contigs (Table 2). The assembly exhibited a GC content of 40.67%. Of these, 136.24 Mb (86.59%) were successfully anchored to 6 pseudo-chromosomes (Fig. 2). BUSCO analysis indicated a completeness score of 98.1%, including 97.4% single-copy, 0.7% duplicated, 0.1% fragmented, and 1.8% missing BUSCOs. The mapping rate for Illumina reads was 95.15%, while PacBio reads had a rate of 99.21%.

Genomic heatmap. Chromosome-level heatmap of Drosophila suzukii, with individual chromosome outlined in blue.
Genome annotation
First, RepeatModeler v2.0.432 was utilized, with an LTR search process, to creat a de novo repeat library based on the structural features of repetitive sequences and ab initio predictions to D. suzukii. The resulting repeat library was combined with Dfam 3.533 and RepBase-2018102634 to generate a comprehensive repeat database. RepeatMasker v4.1.435 was used to predict repeat sequences based on the constructed database. The results revealed 147,215 (27.39 Mb) repetitive sequences, representing 17.41% of the genome. The categories of repetitive sequences with more prevalent were LTR transposons (5.41%), LINE elements (3.15%), simple repeats (2.03%), DNA transposons (1.81%), satellite (1.21%) and unclassified (0.51%) (Table 3).
Protein-coding genes were annotated using MAKER v3.01.0336, which combined ab initio predictions, transcriptome data, and homologous proteins. Ab initio predictions were generated by BRAKER v2.1.637 and GeMoMa v1.838, integrating transcriptomic and protein evidence. The combined predictions were used as input for MAKER. The alignment of transcriptome data to genome was performed using HISAT2 v2.2.039, generating BAM alignment files. BRAKER trained two ab initio tools, Augustus v3.3.440 and GeneMark-ES/ET/EP 4.68_3.60_lic3441, and integrated arthropod protein sequences from the OrthoDB10 v142 database to improve prediction accuracy. Gene structures were predicted by aligning transcript sequences to the genome. StringTie v2.1.643 was used for reference-based transcriptome assembly, utilizing BAM files generated by HISAT2 v2.2.039. Gene prediction was performed using GeMoMa v1.8, which leveraged protein homology and intron position information, with the parameters set as “GeMoMa.c = 0.4, GeMoMa.p = 10”. Homology-based predictions were conducted using protein sequences and annotation files from closely related species, including Anopheles gambiae (GCF_000005575.2), Aedes aegypti (GCF_002204515.2), Drosophila melanogaster (GCF_000001215.4), Drosophila simulans (GCF_016746395.2), and D. suzukii (GCF_037355615.1). We identified 14,742 protein-coding genes, with an average gene length of 5,323.2 bp (Table 4). Each gene contained an average of 4.4 exons (mean length: 474.8 bp), 3.4 introns (mean length: 1,161.5 bp), and 4.2 coding sequences (mean length: 390.2 bp). BUSCO analysis of the predicted protein-coding genes, using insecta_odb10 dataset (n = 1,367), indicated a completeness score of 98.0%.
Gene function annotation was compared against the UniProtKB (SwissProt + TrEMBL) database using Diamond v2.0.11.14944 (‘–very-sensitive -e 1e-5’). Signaling pathways (KEGG, Reactome), gene Ontology (GO) terms, protein domains, and other functional annotations were identified using InterProScan 5.53–87.045 to search Pfam46, SMART47, Superfamily48, and CDD49 databases. EggNOG-mapper v2.1.550 was utilized to query the eggNOG v5.051 database. The final functional annotations were produced by integrating the results from all analyses. Out of the total genes, 14,236 (96.57%) were matched to records in the UniProtKB database, and protein domains were identified for 11,559 genes using InterProScan. Combined InterProScan and eggNOG-mapper results identified 11,018 GO terms and 4,816 KEGG pathways, providing annotations for 10,239 genes with GO terms, 7,816 with KEGG KO terms, 2,730 with enzyme codes, 4,816 with KEGG pathways, and 11,949 with COG functional categories (Table 4). Genomic characteristics were visualized using TBtools v1.09876952 (Fig. 3).

Genomic features of Drosophila suzukii. Each circle from inside to outside represents simple repeats (Simple), long terminal repeats (LTR), long (LINE) and short (SINE) interspersed nuclear elements, DNA transposons density (DNA), GC content (GC), gene density (GENE), and chromosome length (Chr).
Non-coding RNA sequences were identified by comparing against Rfam v14.10 database53 using Infernal v1.1.454. Transfer RNAs were identified using tRNAscan-SE v2.0.955, and low-confidence tRNAs were filtered using the “EukHighConfidenceFilter” script. A total of 726 non-coding RNAs were identified, including 112 ribosomal RNAs, 104 microRNAs, 125 small nuclear RNAs, 327 transfer RNAs, 2 ribozymes and others. Small nuclear RNAs included 34 spliceosomal RNAs, 3 minor spliceosomal RNAs, 8 C/D box snoRNAs, and 30 HCA-box snoRNAs (Table S2).
Chromosome synteny
MMseqs2 was utilized to investigate interspecific chromosomal evolution between D. suzukii and D. melanogaster, applying an e-value threshold of 1e-5. Syntenic blocks were identified using MCScanX56 with the parameter ‘ -s 5’, requiring a minimum of five genes per block to define collinearity. Visualization of the syntenic relationships was performed using TBtools. Comparative genomic analysis revealed substantial interchromosomal alignment between D. suzukii and D. melanogaster, with most chromosomes exhibiting a high degree of shared features. However, instances of chromosomal fusion and fragmentation were also observed. There are also interchromosomal inversion events, primarily involving Dmel4 and Dsuz4 (Fig. 4).

Chromosomal synteny blocks among Drosophila suzukii and Drosophila melanogaster.
Compared to the previous genome version (Genbank: GCA_043229965.1), the genome we obtained exhibits a higher chromosome anchoring rate (Table 5). Chromosome synteny analysis revealed that chromosome fusions were present in the previous version (Fig. S1), likely caused by assembly errors.
Data Records
Raw PacBio, Illumina, Hi-C, and transcriptome sequencing data of D. suzukii have been deposited in the National Center for Biotechnology Information (NCBI) with accession numbers SRP55537957. The assembled genome has been submitted to the NCBI assembly with the accession number JBNGIB000000000.158. The results of annotation for repeated sequences, gene structure, and functional prediction have been made available in Figshare59.
Technical Validation
Two methods were used to evaluate the completeness of the genome assembly. First, BUSCO v5.0.460 with the arthropod reference gene set (n = 1,367) were used to assess the genome assembly. The final assembly achieved a BUSCO completeness score of 98.1%. Second, assembly accuracy was evaluated by calculating the mapping rate. The mapping rate of PacBio reads was 99.21%, while that of Illumina reads was 95.15%.
Code availability
No specific script was used in this work. All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatic software.
References
Tait, G. et al. Drosophila suzukii (Diptera: Drosophilidae): A Decade of Research Towards a Sustainable Integrated Pest Management Program. J. Econ. Entomol. 114, 1950–1974 (2021).
Asplen, M. K. et al. Invasion biology of spotted wing Drosophila (Drosophila suzukii): a global perspective and future priorities. J. Pest Sci. 88, 469–494 (2015).
Atallah, J., Teixeira, L., Salazar, R., Zaragoza, G. & Kopp, A. The making of a pest: the evolution of a fruit-penetrating ovipositor in Drosophila suzukii and related species. Proc. Biol. Sci. 281, 20132840 (2014).
Lee, J. C. et al. The susceptibility of small fruits and cherries to the spotted-wing drosophila, Drosophila suzukii. Pest Manag. Sci. 67, 1358–1367 (2011).
Cini, A., Ioriatti, C. & Anfora, G. A review of the invasion of Drosophila suzukii in Europe and a draft research agenda for integrated pest management. Bull. Insectol. 65, 149–160 (2012).
Little, C. M., Chapman, T. W. & Hillier, N. K. Plasticity Is Key to Success of Drosophila suzukii (Diptera: Drosophilidae) Invasion. J. Insect Sci. 20 (2020).
Matsumura, S. 6000 illustrated insects of the Japanese Empire. (Insecta Matsumurana Tokyo 1931).
Peng, F. T. On some species of Drosophila from China. Anno. Zool. Jpn. 16, 20–27 (1937).
Kaneshiro, K. Y. Drosophila (Sophophora) suzukii (Matsumura). Proc. Hawaiian Entomol. Soc. 24, 179 (1983).
Hauser, M. A historic account of the invasion of Drosophila suzukii (Matsumura) (Diptera: Drosophilidae) in the continental United States, with remarks on their identification. Pest Manag. Sci. 67, 1352–1357 (2011).
Walsh, D. B., Bolda, M., Goodhue, R. & Zalom, F. Drosophila suzukii (Diptera: Drosophilidae): Invasive Pest of Ripening Soft Fruit Expanding its Geographic Range and Damage Potential. J. of Integr. Pest Manag. 2, G1–G7 (2011).
Goodhue, R. E., Bolda, M., Farnsworth, D., Williams, J. C. & Zalom, F. G. Spotted wing drosophila infestation of California strawberries and raspberries: economic analysis of potential revenue losses and control costs. Pest Manag. Sci. 67, 1396–1402 (2011).
Rota-Stabelli, O. et al. Distinct genotypes and phenotypes in European and American strains of Drosophila suzukii: implications for biology and management of an invasive organism. J. Pest Sci. 93, 77–89 (2020).
Lee, J. C. et al. In Focus: Spotted wing drosophila, Drosophila suzukii, across perspectives. Pest Manag. Sci. 67, 1349–1351 (2011).
Deprá, M., Poppe, J. L., Schmitz, H. J., De Toni, D. C. & Valente, V. L. S. The first records of the invasive pest Drosophila suzukii in the South American continent. J. Pest Sci. 87, 379–383 (2014).
Andreazza, F. et al. Drosophila suzukii in Southern Neotropical Region: Current Status and Future Perspectives. Neotrop. Entomol. 46, 591–605 (2017).
Hassani, I. M., Behrman, E. L., Prigent, S. R., Gidaszewski, N. & Yassin, A. First occurrence of the pest Drosophila suzukii (Diptera: Drosophilidae) in the Comoros Archipelago (Western Indian Ocean). Afr. Entomol. 28, 78 (2020).
Gisell, G., Mary, A. L. & Goni, B. Drosophila suzukii (Matsumura) found in Uruguay. Drosophila Information Service 98, 103–107 (2015).
Buonocore Biancheri, M. J. & Kirschbaum, D. S. Drosophila suzukii in Argentina: State of the Art and Further Perspectives. Neotrop. Entomol. 53, 1–17 (2024).
Boughdad, A. et al. First record of the invasive spotted wing Drosophila infesting berry crops in Africa. J. Pest Sci. 94, 261–271 (2021).
EPPO Global Database. Drosophila suzukii (DROSSU)[World distribution]| EPPO Global Database (2025).
Dos Santos, L. A. et al. Global potential distribution of Drosophila suzukii (Diptera, Drosophilidae). PloS one 12, e0174318 (2017).
Weeks, A. R., van Opijnen, T. & Breeuwer, J. A. J. AFLP Fingerprinting for Assessing Intraspecific Variation and Genome Mapping in Mites. Exp. Appl. Acarol. 24, 775–793 (2000).
Bushnell, B. BBtools. Retrieved from https://sourceforge.net/projects/bbmap/ (2014).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Steinegger, M. & Soding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457 (2020).
Hubley, R. et al. The Dfam database of repetitive DNA families. Nucleic Acids Res. 44, D81–89 (2016).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Smit, A. F. A., Hubley, R., & Green, P. RepeatMasker Open-4.0. Retrieved from http://www.repeatmasker.org (2013-2015).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M. & Stanke, M. BRAKER1: Unsupervised RNA-Seq-Based Genome Annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32, 767–769 (2016).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods Mol. Biol. 1962, 161–177 (2019).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–312 (2004).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom. Bioinform. 2, lqaa026 (2020).
Kriventseva, E. V. et al. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47, D807–D811 (2019).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Finn, R. D. et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 (2017).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Letunic, I. & Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496 (2018).
Wilson, D. et al. SUPERFAMILY–sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 37, D380–386 (2009).
Marchler-Bauer, A. et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203 (2017).
Huerta-Cepas, J. et al. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Chen, C. et al. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol Plant 13, 1194–1202 (2020).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–124 (2005).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol. 1962, 1–14 (2019).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
NCBI Sequence reas archive https://identifiers.org/ncbi/insdc.sra:SRP555379 (2025).
Drosophila suzukii whole genome shotgun sequencing project. Genbank https://identifiers.org/ncbi/insdc:JBNGIB000000000 (2025).
Wang, R. Genome annotation of Drosophila suzukii. Figshare. Dataset. https://doi.org/10.6084/m9.figshare.28138766 (2025).
Waterhouse, R. M. et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
Acknowledgements
This work was supported by National Natural Science Foundation of China (32202313), Taishan Scholars Program of Shandong Province (tsqn202312293).
Author information
Authors and Affiliations
Contributions
Y.Z., R.W., Y.L. and L.Z. contributed to the research design. H.G., S.Z., X.D. and Z.Y. collected the samples., R.W., Y.L. and C.W. analyzed the data. L.S. and H.C. contributed to data quality control. R.W., Y.L. and Z.Y. wrote the draft manuscript and revised the manuscript. All co-authors contributed to this manuscript and approved it.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, R., Gao, H., Zhao, S. et al. Chromosomal-level genome assembly of Drosophila suzukii Matsumura (Diptera: Drosophilidae). Sci Data 12, 1600 (2025). https://doi.org/10.1038/s41597-025-05890-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05890-0
