
Abstract
Hippophae neurocarpa (H. neurocarpa) is a deciduous shrub from the Elaeagnaceae family, widely distributed in the Qinghai-Tibet Plateau. It possesses significant ecological and economic value, while also being rich in nutrients and offering various health benefits. In this study, we successfully assembled the chromosome-level genome of H. neurocarpa utilizing Illumina sequencing, Nanopore sequencing, and chromosome conformation capture techniques. The total genome size is 682.80 Mb, with an N50 length of 62.17 Mb. We successfully anchored 669.20 Mb to 12 chromosomes, achieving a mapping rate of 98.0%. Furthermore, using RepeatMasker, we annotated repetitive sequences, revealing that 56.27% of the genome consists of repetitive elements, with long terminal repeats (LTRs) accounting for 36.26% of the total genome. We also annotated gene structures using de novo, homology, and transcriptome assembly strategies, predicting 36844 protein-coding genes, of which 32952 genes had successful functional annotations. Lastly, we employed various strategies for quality evaluation, with a Benchmarking Universal Single-Copy Orthologs (BUSCO) completeness score reaching 97.6% and a Long Terminal Repeat (LTR) Retrotransposon Assembly Index (LAI) of 11.61, the mapping rate further indicating high assembly quality. This study provides valuable genomic data for future research on H. neurocarpa and the broader sea buckthorn genus.
Similar content being viewed by others

Background & Summary
Hippophae neurocarpa (H. neurocarpa) is a member of the Sea buckthorn genus (Hippophae) within the Elaeagnaceae family1. All sea buckthorn species are diploid, with a chromosome count of 2n = 242,3,4. Their distribution spans temperate regions of Eurasia4. These plants exhibit drought tolerance and resilience, and possess root systems capable of forming nitrogen-fixing nodules, which contribute to soil enhancement5. Moreover, Hippophae species demonstrate robust ecological adaptability and stress resistance, playing a vital role in ecological preservation6,7. H. neurocarpa represents one of the most recently diverged and evolutionarily advanced species within this group3,8. As a result of Quaternary climatic fluctuations, natural populations of H. neurocarpa are predominantly found in high-altitude regions of the Qinghai-Tibet Plateau9. This shrub species, endemic to the eastern Qinghai-Tibet Plateau, favors moist environments and exhibits sensitivity to precipitation variations10. The cylindrical fruits of H. neurocarpa are abundant in bioactive compounds, including vitamins A, C, E, K, and P, rendering them a valuable resource in green foods and traditional medicine, with considerable research potential11,12.
Previous studies have extensively investigated the morphological characteristics, ecological traits, and origins of H. neurocarpa. The chloroplast genome of H. neurocarpa has been sequenced, and comparative chloroplast genomics and phylogenetic analyses have been conducted with other species in the Hippophae genus12,13. However, its genetic and evolutionary relationships remain unclear due to the absence of a complete genome sequence. In this study, we employed multiple sequencing technologies to achieve a high-quality genome assembly of H. neurocarpa. We anticipate that this research will not only elucidate the basic characteristics of the H. neurocarpa genome but also enhance the understanding of its adaptability and medicinal properties at the molecular level. Furthermore, these data will provide a robust foundation for future molecular, ecological, and economic research on the entire Hippophae genus.
Methods
Plant materials and genome sequencing
To examine the genomic features of H. neurocarpa, we collected samples from the Tibet Autonomous Region (N 31°14′10″, E 96°36′14″) and extracted high-quality genomic DNA from the leaves using the cetyl trimethylammonium bromide (CTAB) method14. Following the protocols of the MGIEasy Universal DNA Library Preparation Kit, we constructed DNA libraries. These libraries were subsequently submitted to GrandOmics Bioscience Co, Ltd. for high-throughput sequencing using the DNBSEQ-T7RS platform to generate second-generation sequencing data. To ensure data quality, we employed Fastp v0.23.215 software (fastp -w 8 -n 0 -l 140) to filter the raw sequencing data generated by the Illumina platform, ultimately obtaining 66.79 Gb of high-quality clean next-generation sequencing (NGS) reads (Table 1). For Oxford Nanopore Technologies (ONT) sequencing, we adhered to the manufacturer’s guidelines, extracting DNA with a Grandomics kit, and utilized the Blue Pippin and Pippin HT systems to carefully select long DNA fragments. Following damage repair, end repair, and A-tailing, sequencing adapters were successfully ligated, and the library concentration was accurately measured using a Qubit® 3.0 fluorometer. Finally, sequencing was conducted on the Nanopore PromethION platform, yielding genome data that comprised 94.90 Gb of Nanopore long reads (Table 2).
Hi-C Sequencing
Hi-C sequencing is a high-throughput chromosome conformation capture technique that enables the analysis of the three-dimensional structure of chromosomes across the entire genome16. Following established research protocols, genomic DNA was extracted from young leaves of H. neurocarpa and sequenced using the DNBSEQ-T7RS platform. High-quality Hi-C samples were prepared through processes including formaldehyde cross-linking, cell lysis, chromatin digestion, and biotin labeling. The DNA quality was assessed, and high-quality DNA was retained for the standard library construction process. PCR conditions were optimized before amplification. Prior to constructing the sequencing library, a “Hi-C fragment ligation quality control test” was performed on the amplified products to ensure data accuracy. The DNBSEQ-T7RS platform ultimately generated 73.40 Gb of high-quality Hi-C data (Table 3).
Genome survey and evaluation
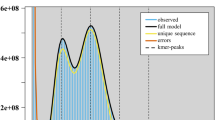
In this study, we conducted K-mer analysis (k-mer = 21) on the clean NGS data, utilizing Jellyfish v2.3.017 (jellyfish count -t 22 -C -m 19 -s 1000 -o)software for processing. Subsequently, we employed GenomeScope v2.018 (genomescope.R -i k21.histo -o k21.gs -k 21)for visualization, precisely estimating the genome size and heterozygosity of H. neurocarpa at 684 Mb and 1.04%, respectively (Fig. 1).

K-mer distribution (K = 21) of Hippophae neurocarpa genome analyzed using GenomeScope 2.
De Novo genome assembly
This study integrated advanced genome sequencing technologies, including Nanopore long reads, MGI short reads, and Hi-C reads, to perform a comprehensive and detailed de novo assembly of the H. neurocarpa genome using clean Illumina and Nanopore data. Initially, NextDenovo (v2.5.0) (https://github.com/Nextomics/NextDenovo) was utilized to construct an initial genome framework based on the Nanopore data. Subsequently, the preliminary assembly results were optimized and corrected using clean Illumina data with NextPolish (v1.4.1)19 (nextpolish2 -t 5), significantly enhancing assembly accuracy. To eliminate potential redundant sequences in the genome, Purge-dups (v1.2.5)20 (purge_dups -2 -T -c)was employed to construct a more refined and accurate chromosome-level genome assembly. Following this, Hi-C data were validated with HiCPro (v3.1.0)21 (hicpro -i reads -o out -c), and detailed manual inspection and fine-tuning were conducted using Juicebox assembly tools22 and high-quality Hippophae salicifolia as a reference genome23. This process included cluster optimization, sequence reordering, and orientation corrections, ensuring that each step met rigorous standards.
Building upon this foundation, we employed 3D-DNA24 tools (run-3ddna-pipeline.sh -r -c) to accurately anchor the contigs to chromosomes, resulting in a final chromosome-level assembly. The total genome size reached 682.80 Mb, comprising 38 scaffolds with an N50 length of 62.17 Mb. The 12 main chromosomes encompassed over 98.0% of the entire genome (Table 4). Furthermore, we utilized BUSCO (v5.4.5)25 (busco -i -o -c 70 -m geno -l) to comprehensively assess genome quality, with results indicating a genome completeness of 97.6%, confirming the high quality and completeness of the assembly. Concurrently, PlantLAI26-based (https://bioinformatics.um6p.ma/PlantLAI/) evaluation of long terminal repeat (LTR) retrotransposons yielded an assembly index (LAI) of 11.61, indicating that the genome assembly is of reference quality. To visually represent the distribution of genome elements, we employed ShinyCircos v2.027 (https://venyao.xyz/shinyCircos/) to generate a Circos plot, clearly illustrating the structural features of the genome (Fig. 3). Additionally, the whole-genome Hi-C heatmap generated using HiCExplorer28 (hicPlotMatrix–title–matrix–dpi–colorMap–log1p–fontsize 8–rotationX 30 –outFileName) vividly revealed interchromosomal interaction patterns, providing robust data support and visualization tools for further exploration of the genome’s complex structure and functional characteristics (Fig. 2).

Heatmap of genome-wide Hi-C data of Hippophae neurocarpa chromosomes.
Annotation of repetitive sequences
In annotating the repetitive sequences of the sea buckthorn genome, we utilized a comprehensive tool—Extensive de novo TE Annotator (EDTA, v2.1.2)29 (edta–genome–sensitive 1–threads)—to integrate results from multiple prediction tools. Initially, the transposable element (TE) library obtained from EDTA was further classified using TEsorter (v1.33)30 (TEsorter -db rexdb-plant -p 56 -pre), specifically reclassifying elements in the “LTR-unknown” category. Subsequently, we conducted an in-depth classification analysis of these reclassified elements using DeepTE31 (deepTE_domain.py -d -s -i -d). Ultimately, we merged the results from the three independent TE databases and employed RepeatMasker (v4.1.2)32 (repeatmasker -pa 14 -s -xsmall -lib), which defaults to the RepBase RepeatMasker Edition (RMB, 20181026) version of the repeat sequence database, to identify homologous sequences in the total TE database.
The analysis revealed that repetitive elements comprise 56.27% of the sea buckthorn genome, with LTRs accounting for 36.26% and terminal inverted repeats (TIRs) accounting for 12.21% (Table 5, Fig. 3). Furthermore, we used the MegaLTR33 (MegaLTR.sh -A 3 -F -G -T -P results -l 100 -L 7000 -d 1000 -D 15000 -S 85 -M 20 -B rexdb -C 20 -V 0.001 -Q 80-80-80 -E rexdb -R 0.000000015 -U 5000 -X 5000 -W 1000000 -N 12 -t 104) software to conduct a more refined classification of LTRs into superfamilies and lineages. We found that the LTR retrotransposons in H. neurocarpa are categorized into four superfamilies, specifically Ty1-copia, Ty3-gypsy, BARE-2, and TR-GAG. Additionally, we identified 13 distinct lineages, which include Ale, Tork, Athila, Ivana, Galadriel, Ikeros, SIRE, TAR, CRM, Reina, Crm, and Reina (Tables 6, 7).

Circos plot illustrating the genomic landscape of Hippophae neurocarpa. (a) Gene density. (b) Repeat sequence density. (c) Copia element density. (d) Gypsy element density. (e) GC content. (f) Interspecies collinearity.
In addition, we employed the MISA (v2.1)34 (misa.pl genome.fa) to identify SSR loci across the entire genome of Hippophae neurocarpa. The analysis identified 205,053 SSR loci across 12 sequences. All examined sequences contained multiple microsatellite loci, with mononucleotide and dinucleotide repeats being predominant, accounting for 49.1% and 37.1% of the total SSR loci, respectively. The frequency of SSRs decreased significantly with increasing repeat unit length (3–6 nucleotides). Notably, 20,473 compound microsatellites were detected, representing 10% of the total SSRs, indicating that a considerable proportion of microsatellites exist in tandem arrangements (Table 8).
Annotation of protein-coding gene structure
For the structural annotation of protein-coding genes, we employed three prediction methods: de novo prediction, homology protein sequence alignment, and RNA-seq data analysis. Initially, we performed soft masking of repetitive sequences in the genome using RepeatMasker. Subsequently, we annotated the masked genome using BRAKER (v3.0.8)35 software (braker.pl–genome–bam–prot_seq–species–threads 56–workingdir–PROTHINT_PATH–TSEBRA_PATH). The prediction results were then merged using TSEBRA36 software (tsebra.py–cfg–gtf–keep_gtf–hintfiles–out). Finally, we integrated the annotation files using MAKER (v3.01.04)37 (mpirun -n -R–ignore_nfs_tmp -TMP) and EVidenceModeler (v1.1.1)38 to obtain non-redundant gene models, resulting in a GFF3 file that includes the locations of genes, coding sequences, proteins, and mRNA. In total, we predicted 36,844 protein-coding genes, with gene lengths ranging from 155 to 185,811 bp.
Non-coding region annotation
Non-coding RNAs in H. neurocarpa were identified using the Infernal (v1.1.4)39 (infernal-tblout2gff.pl–cmscan–fmt2–desc) search tool with the Rfam40 database, employing default parameters. This analysis revealed 5917 non-coding RNAs spanning 817623 bp, comprising 724 transfer RNAs (53476 bp), 4194 small nucleolar RNAs (440956 bp), 607 ribosomal RNAs (270544 bp), 84 spliceosomal nuclear RNAs (11934 bp), 182 microRNAs (23063 bp), and 126 other RNA types totaling 17650 bp. It is particularly noteworthy that snoRNAs dominate with 4,194 loci (accounting for 70.9%), primarily involved in the modification and processing of rRNAs and tRNAs. The 724 tRNAs and 607 rRNAs collectively maintain the proper functioning of the protein synthesis system, while 182 miRNAs participate in fine-tuning gene expression through post-transcriptional regulation. Additionally, 84 spliceosomal RNAs are responsible for precise pre-mRNA splicing, along with the identification of 126 ncRNAs whose functions remain to be elucidated. These ncRNAs work synergistically to play crucial roles in key biological processes including transcriptional regulation, protein synthesis, and RNA processing (Table 9).
Functional annotation
To enhance the functional annotation of predicted genes, we conducted a homology search using the BLASTP41 tool in conjunction with multiple public databases accessible through the Baimaike website (https://international.biocloud.net/zh/user/login), employing an e-value threshold of 1e−10. The databases utilized included non-redundant database (NR), Swissprot42, TrEMBL43, KOG, Gene Ontology (GO)44, KEGG45, and COG46. The analysis resulted in functional annotations for 89.44% of the genes. Specifically, the annotation percentages for individual databases were as follows: NR (89.22%), TrEMBL (92.01%), Swissprot (61.98%), KOG (50.90%), KEGG (15.22%), GO (5.43%), and COG (30.96%) (Table 10). To conclude, we assessed the annotation results using the OMArk47 website (https://omark.omabrowser.org/), and the genome annotation assessment revealed that among the 10,551 conserved orthologous gene groups (HOGs), the completeness rate reached 96.21%, with only 3.79% missing, demonstrating highly complete and reliable annotation results (Table 11).
Furthermore, we conducted systematic GO functional classification, KEGG pathway enrichment analysis, and gene family distribution of the annotated results. Out of 2001 annotated genes, 5606 were annotated by KEGG (Table 10). The GO analysis indicated a predominance of cellular components, molecular functions, and biological processes, highlighting biological processes (Fig. 4). KEGG analysis showed significant enrichment in metabolism and Genetic Information Processing pathways (Fig. 5). The identified gene families, such as RVT_2, UBN2, and Lipase_GDSL, suggest active transposon activity, essential protein functions, and complex metabolic pathways in Ribesia sandwedge. All functionally annotated genes were classified into 2,071 gene families, with predominant enrichment observed in families such as RVT_2, UBN2, NAM, among others, suggesting the potential presence of active transposable element activity, important intracellular protein functions, and complex metabolic pathways in H. neurocarpa.

GO illustrating the functional annotation of Hippophae neurocarpa.

KEGG illustrating the functional annotation of Hippophae neurocarpa.
Data Records
The raw sequencing data are publicly available in the Genome Sequence Archive (GSA) in National Data Center (https://ngdc.cncb.ac.cn/gsa) under the number CRA02068748. The genome assembly sequences and annotation files, including Gene Ontology (GO) annotation statistics, KEGG pathway analysis results, and gene family classification statistics, have been deposited in Figshare49 and NCBl GenBank database50.
Technical Validation
The completeness of the non-redundant draft genome was evaluated using Benchmarking Universal Single-Copy Orthologs (BUSCO)24 with the embryophyta odb10 dataset, which consists of 1614 single copy genes with the default parameters. Revealing that 97.6% of these genes exhibited complete coverage. Among them, 97.6% were complete, 89.0% were complete and single-copy (Table 12).
Data availability
The raw sequencing data are publicly available in the Genome Sequence Archive (GSA) in National Data Center under the number CRA02068748 (https:/ngdc.cncb.ac.cn/gsa/browse/CRA020687). The genome assembly sequences and annotation files, including Gene Ontology (GO) annotation statistics, KEGG pathway analysis results, and gene family classification statistics, have been deposited in Figshare49 (https://doi.org/10.6084/m9.figshare.27890145.v6) and NCBl GenBank database50 (https://identifiers.org/ncbi/insdc:JBPJAO000000000).
Code availability
No specific code was developed in this work.
References
He, S. W. L. T. N. Hippophaë neurocarpa. Acta Phytotax 16(2), 107 (1978).
Corriveau, J. L. & Coleman, A. W. Rapid screening method to detect potential biparental inheritance of plastid DNA and results for over 200 angiosperm species. Am. J. Bot. 75, 1443–1458 (1988).
Zhao, J., Zhang, Z., Zhou, H., Bai, Z. & Sun, K. The Study on Sea Buckthorn (Genus Hippophae L.) Fruit Reveals Cell Division and Cell Expansion to Promote Morphogenesis. Plants 12, 1005 (2023).
Yang, X. et al. Chromosome-level genome assembly of Hippophae rhamnoides variety. Sci.Data 11, 776, https://doi.org/10.1038/s41597-024-03549-w (2024).
Zhou, X. et al. Rhizospheric fungi and their link with the nitrogen-fixing Frankia harbored in host plant Hippophae rhamnoides L. J. Basic Microbiol. 57, 1055–1064, https://doi.org/10.1002/jobm.201700312 (2017).
Chen, M. et al. Chromosome-level genome assembly of Hippophae gyantsensis. Sci.Data 11, 126, https://doi.org/10.1038/s41597-024-02909-w (2024).
Kortesniemi, M., Sinkkonen, J., Yang, B. & Kallio, H. NMR metabolomics demonstrates phenotypic plasticity of sea buckthorn (Hippophaë rhamnoides) berries with respect to growth conditions in Finland and Canada. Food Chem 219, 139–147, https://doi.org/10.1016/j.foodchem.2016.09.125 (2017).
LI, N. et al. Genetic diversity and genetic structure of the northern margin populations of Hippophae neurocarpa. Guihaia 36, 557–563, https://doi.org/10.11931/guihaia.gxzw201511006 (2016).
Meng, L.-H., Yang, H., Wu, G. & Wang, Y.-J. Phylogeography of Hippophae neurocarpa (Elaeagnaceae) inferred from the chloroplast DNA trnL-F sequence variation. J SYST EVOL 46, 32–40 (2007).
Kou, Y.-X., Wu, Y.-X., Jia, D.-R., Li, Z.-H. & Wang, Y.-J. Range expansion, genetic differentiation, and phenotypic adaption of Hippophaë neurocarpa (Elaeagnaceae) on the Qinghai–Tibet Plateau. J SYST EVOL 52, 303–312, https://doi.org/10.1111/jse.12063 (2014).
Meng, D. et al. An insight into the association between volatile and other active components of sea buckthorn berries in barren soils of western China. Food and Humanity 2, 100293, https://doi.org/10.1016/j.foohum.2024.100293 (2024).
Zhou, W., Hu, N., Dong, Q., Wang, H. & Wang, Y. Complete chloroplast genome sequences of Hippophae neurocarpa. Mitochondrial DNA Part B-Resources 4, 2048–2049, https://doi.org/10.1080/23802359.2019.1617077 (2019).
Li, X., Liu, L. E., Li, H., Li, J. & Zhou, W. Chloroplast Genomes and Phylogenetic Analysis of Hippophae tibetana and Hippophae neurocarpa. Genomics and Applied Biology 43, 767–787 (2024).
Murray, M. G. & Thompson, W. F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8, 4321–4325, https://doi.org/10.1093/nar/8.19.4321 (1980).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Belton, J. M. et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276, https://doi.org/10.1016/j.ymeth.2012.05.001 (2012).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255, https://doi.org/10.1093/bioinformatics/btz891 (2019).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259, https://doi.org/10.1186/s13059-015-0831-x (2015).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Chen, M. et al. The genome of Hippophae salicifolia provides new insights into the sexual differentiation of sea buckthorn, GigaScience, Volume 14, https://doi.org/10.1093/gigascience/giaf046 (2025).
Zhang, Y., Xiong, Y. & Xiao, Y. 3dDNA: A Computational Method of Building DNA 3D Structures. Molecules 27, https://doi.org/10.3390/molecules27185936 (2022).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Mokhtar, M. M., Abd-Elhalim, H. M. & El Allali, A. A large-scale assessment of the quality of plant genome assemblies using the LTR assembly index. AoB PLANTS 15, https://doi.org/10.1093/aobpla/plad015 (2023).
Wang, Y. et al. shinyCircos-V2.0: Leveraging the creation of Circos plot with enhanced usability and advanced features. iMeta 2, https://doi.org/10.1002/imt2.109 (2023).
Wolff, J. et al. Galaxy HiCExplorer 3: a web server for reproducible Hi-C, capture Hi-C and single-cell Hi-C data analysis, quality control and visualization. Nucleic Acids Res. 48, W177–W184, https://doi.org/10.1093/nar/gkaa220 (2020).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275, https://doi.org/10.1186/s13059-019-1905-y (2019).
Zhang, R.-G. et al. TEsorter: An accurate and fast method to classify LTR-retrotransposons in plant genomes. Hortic. Res. 9, https://doi.org/10.1093/hr/uhac017 (2022).
Yan, H., Bombarely, A. & Li, S. DeepTE: a computational method for de novo classification of transposons with convolutional neural network. Bioinformatics 36, 4269–4275, https://doi.org/10.1093/bioinformatics/btaa519 (2020).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358, https://doi.org/10.1093/bioinformatics/bti10 (2005).
Mokhtar, M. M. & El Allali, A. MegaLTR: a web server and standalone pipeline for detecting and annotating LTR-retrotransposons in plant genomes. Frontiers in plant science 14, 1237426, https://doi.org/10.3389/fpls.2023.1237426 (2023).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585 (2017). 10.1093/bioinformatics/btx198%JBioinformatics.
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 3, https://doi.org/10.1093/nargab/lqaa108 (2021).
Gabriel, L., Hoff, K. J., Brůna, T., Borodovsky, M. & Stanke, M. TSEBRA: transcript selector for BRAKER. BMC Bioinform. 22, 566, https://doi.org/10.1186/s12859-021-04482-0 (2021).
Campbell, M. S., Holt, C., Moore, B. & Yandell, M. Genome Annotation and Curation Using MAKER and MAKER-P. Curr. Protoc. Bioinformatics 48, 4.11.11–14.11.39, https://doi.org/10.1002/0471250953.bi0411s48 (2014).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013). 10.1093/bioinformatics/btt509%JBioinformatics.
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124, https://doi.org/10.1093/nar/gki081 (2005).
Jacob, A., Lancaster, J., Buhler, J., Harris, B. & Chamberlain, R. D. Mercury BLASTP: Accelerating Protein Sequence Alignment. TRETS 1, 9, https://doi.org/10.1145/1371579.1371581 (2008).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370, https://doi.org/10.1093/nar/gkg095 (2003).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48, https://doi.org/10.1093/nar/28.1.45 (2000).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29, https://doi.org/10.1038/75556 (2000).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462, https://doi.org/10.1093/nar/gkv1070 (2015).
Tatusov, R. L., Galperin, M. Y., Natale, D. A. & Koonin, E. V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36, https://doi.org/10.1093/nar/28.1.33 (2000).
Nevers, Y. et al. Quality assessment of gene repertoire annotations with OMArk. Nat. Biotechnol. 43, 124–133 (2025).
CNCB-NGDC Genome Sequence Archive. https://ngdc.cncb.ac.cn/gsa/browse/CRA020687 (2025).
Tian, Z. data for Hippophae neurocarpa genome data. figshare. Dataset. https://doi.org/10.6084/m9.figshare.27890145.v6 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBPJAO000000000 (2025).
Acknowledgements
This research was supported by the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (2019QZKK0502), Key Research and Development Program for Bureau of Science and Technology of Xizang Autonomous Region (XZ202401ZY0006), Key Research, Development and Transformation Program for Shigatse City Bureau of Science and Technology (RKZ2023ZY-03).
Author information
Authors and Affiliations
Contributions
Yunqiang Yang conceived the study and supervised the project. Ziyue Tian wrote the manuscript and participated in the data analysis. Xingyu Yang, Danni Yang, Jian Huang, Shihai Yang, Yunqiang Yang collected the samples, performed the figures drawing and upload the data. All authors have read, revised, and approved the final manuscript for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declear no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tian, Z., Yang, X., Yang, D. et al. Chromosome-level genome assembly of Hippophae neurocarpa. Sci Data 12, 1743 (2025). https://doi.org/10.1038/s41597-025-06022-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06022-4
