
Abstract
Fraxinus mandshurica Rupr, an important timber species, is widely distributed throughout northeastern China. In our study, we assembled the telomere-to-telomere (T2T) genome of F. mandshurica, generated by using Illumina short-read, PacBio HiFi long-read and Hi-C sequencing. The genome size was 781.40 Mb and the scaffold N50 length was 34.29 Mb. 39 contigs were reliably scaffolded into 23 T2T chromosomes. More than 98.70% of the BUSCO genes from the embryophyta_odb12 dataset were successfully hit in the genome. We found that 61.01% of the sequences were repetitive, and 35,009 protein-coding genes were annotated, more than 99.22% were successfully functionally detected. Overall, this T2T-level genome assembly provides a valuable genomic platform that will facilitate studies of evolutionary biology, functional genomics, and molecular breeding in F. mandshurica.
Similar content being viewed by others


Background & Summary
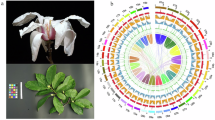
Fraxinus mandshurica Rupr, belongs to the Oleaceae family and Fraxinus genus, is both economically and ecologically significant. It is mainly distributed in Northeast China, with smaller populations found in Northwest China, Russia, Japan, and North Korea1. F. mandshurica is a deciduous tree with pinnate leaves bearing ovate leaflets with serrulate margins. Its seeds are samaras with twisted wings extending to the base of the nutlet2 (Fig. 1). Renowned as a valuable timber species, it produces valuable wood that is widely used for furniture, flooring, and handicrafts. Beyond its economic importance, the species holds substantial medicinal value. Its bark, leaves, and other parts have been extensively utilized in traditional Chinese medicine, as well as in the traditional remedies of other Asian countries, such as South Korea and Japan3.

Morphological features of Fraxinus mandshurica Rupr. (A) Seedlings of F. mandshurica used for sequencing. (B) Left – abaxial (upper) surface of F. mandshurica leaf; Right – adaxial (underside) surface. (C) Seed of F. mandshurica.
However, in recent decades, environmental degradation has led to a significant decline in the wild populations of F. mandshurica4. Furthermore, the lack of a high-quality reference genome has posed substantial challenges to conducting in-depth research on this species. Currently, genomes of several Fraxinus species have been published5, including F. americana, F. excelsior and F. pennsylvanica, among others, but the telomere-to-telomere (T2T) genome assembly of F. mandshurica is still lacking. Although transcriptome analyses6, gene family studies7, and structural variations analyses5 in F. mandshurica are gradually increasing, these studies all rely on the availability of a high-quality reference genome. A T2T genome provides a comprehensive view of tandem repeat sequences such as centromeres and telomeres, and enables more accurate and complete prediction of protein-coding genes8. To address these problems, we have constructed a high-quality reference genome for F. mandshurica. This genomic resource will facilitate preservation genetics research and provide insights into the molecular mechanisms underlying the species’ important economic phenotypes, ultimately aiding in its preservation and sustainable utilization.
In this work, we utilized Illumina (144.80 × ), PacBio HiFi (115.34 × ), and Hi-C data (126.10 × ) to construct the T2T genome. High-coverage HiFi sequencing data alone can assemble 19 complete T2T chromosomes without relying on Hi-C scaffolding (Table 1). With the assistance of Hi-C, we further anchored the remaining chromosomes and filled the gaps, resulting in the first T2T genome assembly of F. mandshurica. The final assembly consists of 23 chromosomes, with a total genome size of 781.40 Mb, the contig N50 of 34.29 Mb, and the contig L50 of 10. A total of 35,009 protein-coding genes were predicted in our genome, of which 34,735 were successfully functionally annotated (Fig. 2). A higher sequencing depth can significantly improve the quality of genome assembly, indicating that sufficient data coverage is crucial for obtaining a more accurate and complete genome. Compared with the previously published genome in China National Genomics Data Center (Accession: GWHFDPP00000000.1)9, with a nearly equivalent level of BUSCO completeness, our genome exhibits further enhancements in contig number, contig N50, and L50 relative to assembly GWHFDPP00000000.1, which indicates a higher degree of continuity in the new assembly (Table 2).

Genome characteristics of Fraxinus mandshurica Rupr. (A) A Circos plot illustrating, from the outside inwards: chromosomes (with each tick representing 1 Mb), gene density, repeat sequence density, GC content, and intra-genomic synteny. (B) Schematic representation of chromosome structure. (C) UpSet plot summarizing the functional annotation of the genome.
Methods
Sample collection
F. mandshurica plant from which the samples were tissue-cultured was collected, cultivated, and preserved at the National Forest Seed Base of Jilin Forest Industry Hongshi Forestry Co., Ltd., Huadian, China. Genomic DNA was extracted from three stages of young leaves with varying degrees of tenderness: the most tender leaf (from the middle of the apical part), a moderately tender leaf, and a slightly less tender leaf. These were used for Illumina short-read sequencing, PacBio HiFi long-read sequencing (CCS), and Hi-C sequencing. RNA samples were extracted from three different tissues—leaves, bark, and roots—collected at the same developmental stage of the plant. All samples used for DNA and RNA extraction were obtained from the same individual plant.
DNA and RNA sequencing
The phenol/chloroform extraction protocol was applied to fresh leaf tissue to obtain genomic DNA for sequencing library preparation. Short-read sequencing was typically used for genome survey, providing a preliminary understanding of genome characteristics such as size, heterozygosity, and repeat content. Long-read sequencing, on the other hand, is employed for de novo genome assembly due to its ability to span repetitive regions and produce longer contigs. Finally, Hi-C data was utilized to correct and scaffold the contigs.
The integrity of the extracted genomic DNA was assessed using agarose gel electrophoresis. A paired-end sequencing library with an insert size of 300–400 bp was constructed and sequenced on the Illumina NovaSeq 6000 platform, generating a total of 113.15 Gb of raw short reads (150 bp in length). Additionally, PacBio HiFi long reads were obtained using the PacBio Sequel II platform in Circular Consensus Sequencing (CCS) mode, which provides both long read lengths and high base-level accuracy. In total, 90 Gb of HiFi sequencing data were generated for genome assembly.
We also performed Hi-C sequencing, the genomic DNA conformation is fixed in cells using paraformaldehyde, followed by cell lysis and treatment of the crosslinked DNA with restriction enzymes to generate sticky ends. The DNA ends are then repaired, during which biotin is introduced to label the oligonucleotide ends. Subsequently, DNA fragments are ligated using DNA ligase, and protein digestion is performed to reverse the DNA-protein crosslinking. The DNA is then purified and randomly fragmented into 300–500 bp fragments, and the biotin-labeled DNA is captured using streptavidin magnetic beads and prepared for short-read library sequencing. Finally, the raw data amounted to 98.54 GB, consisting of a total of 656,944,166 reads.
Total RNA was extracted from F. mandshurica leaves, barks, and roots. RNA quality was assessed based on appearance, purity (NanoDrop), concentration (Qubit), and integrity (agarose gel electrophoresis). mRNA was enriched using oligo (dT) magnetic beads, fragmented, and reverse-transcribed to cDNA. After end repair, A-tailing, and adapter ligation, libraries were purified and optionally PCR-amplified. Library quality was evaluated, pooled, and sequenced on the Illumina NovaSeq 6000 platform.
T2T genome assembly
The short reads were quality-controlled and filtered using fastp (v0.23.4)10 to obtain clean data. Based on 97.69 Gb of clean short reads, we used the Kmer-based analysis to estimate genome size, heterozygosity, and repetitive sequence percentage. We counted the number of each 17-mer with GCE (v1.0.0)11. The analysis results revealed that the F. mandshurica genome size was approximately 810 Mb, with an adjusted size of 804 Mb. The heterozygosity rate was 0.82%, and the proportion of repetitive sequences was 55.94%. We were also use Smudgeplot (v0.2.3dev)12 with -k21 -m100 -ci1 -cs10000 to analyze genome structure. The AB-type determined that the most likely ploidy of F. mandshurica is diploid. (Fig. 3).

The k-mer count distribution for the genome size estimation and genomic haplotype analyse.
We used an integrated genome assembly strategy combining hifiasm (v0.25.0)13 with the parameter–telo-m AAACCCT and verkko (v2.2.1)14,15 with the parameter–telomere-motif AAACCCT to assemble HiFi sequencing data, with both tools running in Hi-C mode. The telomere motif used is a common plant telomeric sequence16, and telomeric sequence identification using tidk(v0.2.0)17 confirmed its presence without variation. By comparing the number of telomeres from both assembly tools (Table 2), we selected the hifiasm assembly as the preliminary assembly result. Due to the high heterozygosity and repetitive sequence percentage of the genome, the assembled genome size exceeded our expectations. We employed purge_dups (v1.2.5)18 to remove redundant sequences from the initial assembly, using minimap2 (v2.28)19 to align the raw reads back to the assembled genome. Redundant contigs were identified and removed based on read depth distribution and sequence similarity, resulting in 39 contigs. We then used quarTeT (v1.2.5)20 with the parameter te –c plant to detect telomeric signals, identifying 19 contigs as complete chromosomes with telomeric repeat sequences at both ends.
To generate a complete T2T genome assembly, the clean Hi-C data were aligned to our genome by using Chromap (v0.2.7-r494)21 and scaffolded with YaHS (v1.2.2)22. The resulting scaffolds were manually curated using Juicebox(v1.11.08) (GitHub - aidenlab/Juicebox: Visualization and analysis software for Hi-C data -) to correct misassemblies and improve chromosome-level organization. As a result, the F. mandshurica genome was successfully anchored onto 23 chromosomes. Gaps during the scaffolding process were filled using TGS-GapCloser (v1.2.1)23, with additional gap filling assisted by Verkko-assembled contigs. This process yielded a T2T genome of F. mandshurica comprising 45 telomeres and no gap (Fig. 4).

Hi-C contact map and contig distribution map on chromosomes of the genome. (A)The map shows scaffolded and independently assembled chromosomes at 500 kb resolution in 5 Mb windows. (B) Contig Distribution Map on Chromosomes of the Genome.
Repeat elements identification
Before performing genome annotation, to comprehensively identify and mask repetitive elements in the F. mandshurica genome, we employed a combination of de novo and homology-based approaches. Tandem repeats were first identified using Tandem Repeats Finder (TRF) (v4.09.1)24. For de novo transposable element (TE) discovery, we applied RepeatModeler (v2.0.1)25, which constructs a custom repeat library specific to the genome. In parallel, we utilized EDTA (v2.2.2)26, an integrated pipeline optimized for plant genomes, to annotate and classify TEs with high sensitivity. The custom repeat libraries generated by de novo prediction tools were then used as input for RepeatMasker (v4.1.7)27 to screen the genome for repetitive sequences. For homology-based repeat annotation, we conducted masking with RepeatMasker using both the Dfam28 and Repbase29 libraries, ensuring comprehensive coverage of known repetitive elements. Additionally, RepeatProteinMask was used to detect TEs at the protein level, further improving annotation accuracy. To ensure accuracy and reduce redundancy, we removed overlapping regions among the results obtained from the various methods, retaining only non-redundant repeat annotations. In total, 61.01% of the genome was masked as repetitive (Table 3), a proportion consistent with that reported in currently published Fraxinus genomes5. This indicates that our integrative strategy enabled robust and reliable repeat identification and masking.
Gene annotation
To ensure high-quality and high-fidelity annotation, we employed the EviAnn (v2.0.2)30 software, which performs purely evidence-based annotation rather than relying on gene prediction models. The annotation was based on two sources of evidence: the protein from closely related species and the transcriptome data derived from the same organism. The protein dataset, comprising approximately 335,013 protein sequences from Oleaceae family, was obtained from NCBI31. For transcriptome evidence, to minimize potential gene annotation omissions caused by insufficient transcriptome coverage, we supplemented our own RNA-seq data—generated from root, bark, and leaf tissues—with additional transcriptome datasets derived from other F. mandshurica tissues, including flowers (Accession: SRP513361)32, pollen (Accession: SRP522725)33, and stigmas(Accession: SRP559292)34, which were downloaded from NCBI. All RNA-seq reads were aligned to the reference genome using HISAT2 (v2.2.1)35. Samtools (v1.9)36 was used to change SAM files to BAM format, followed by sorting and merging of the BAM files to generate a coordinated and consolidated alignment dataset. Both protein and transcriptome evidence were then supplied to EviAnn to generate the final gene annotation. A total of 35,009 protein-coding genes and 57,195 transcripts were ultimately predicted, which is consistent with the previously published genome of Fraxinus pennsylvanica37. (Fig. 5A and B).

Structural comparison of the gene models among the Fraxinus mandshurica Rupr and classification of GO and KEGG. (A) Gene element length distribution plot of the gene set. (B) Cumulative gene element length distribution plot of the gene set. (C) GO classification bar chart. (D) KEGG classification bar chart.
To functionally annotate the predicted genes, we first performed sequence similarity searches against major protein databases including NR38, SwissProt39, and TrEMBL39 using DIAMOND (v0.9.25.126)40 (parameters:–max-target-seqs. 1–evalue 1e-5). These alignments allowed for the initial functional characterization of the gene set based on homology. To further investigate potential protein functions, we applied InterProScan (v5.61-93.0)41 and HMMER3 (v3.3.1)42 to identify conserved domains by querying the InterPro43 and Pfam44 databases. The annotated domains were subsequently mapped to Gene Ontology (GO) terms45, and GO enrichment analysis was performed (Fig. 5C). In addition, KofamScan (v1.3.0)46 was used to assign KEGG47 identifiers to the genes, providing pathway-level insights into gene functions (Fig. 5D). Finally, 99.22% of the genes were functionally annotated (Table 4).
We also annotated non-coding RNAs by employing various methods, tRNA sequences were identified using tRNAscan-SE (v1.3.1)48; rRNA sequences were annotated using BLASTN (v2.14.1 + )49 with reference sequences from closely related species due to their high conservation; and miRNA and snRNA sequences were annotated using the INFERNAL (v1.1.5)50 from the Rfam (v14.8)51 (Table 5).
Data Records
The sequencing dataset and genome assembly were deposited in public repositories All raw date has been submitted to the NCBI under the BioProject accession number PRJNA1273044.
The genomic Illumina (SRR33856795), PacBio HiFi (SRR33856794), and Hi-C (SRR33856793) sequencing data were deposited in the NCBI Sequence Read Archive. The transcriptome Illumina sequencing data from leaves (SRR33856792), barks (SRR33856791), and roots (SRR33856790) were also deposited in the NCBI Sequence Read Archive (Accession: SRP590381)52.
The final assembly genome was submitted to NCBI Assembly with accession number GCA_050941835.153. The GFF file was available on Figshare (https://doi.org/10.6084/m9.figshare.29424683.v1)54.
Technical Validation
Identification of telomeric and centromeric regions
Telomeres and centromeres were located using quartet (v1.2.5)20 with the parameters–TE–gene. The TE annotations were derived from the annotation files generated by EDTA during repeat masking, while the gene annotations were obtained by integrating mRNA annotations from EviAnn and non-coding RNA annotations from Infernal. A total of 45 telomeres and 23 centromeres were identified (Table 6). Notably, the only missing telomere was at the end of chromosome 23, where a high density of rRNA annotations was observed. This suggests that the highly repetitive rDNA region may have hindered the complete assembly of the terminal telomeric sequence.
Quality assessment of the genome and proteins
In the raw sequencing data, adapter sequences, low-quality bases, and unsequenced bases (represented by N) can significantly interfere with downstream analysis. Through fastp (v0.23.2)10 filtering and redundancy removal.
To evaluate the quality of the assembled genome, the completeness of the assembled genome was assessed by using BUSCO (v5.8.2)55 and Compleasm (v0.2.7)56 with the embryophyta_odb12. We found that 98.7% and 99.9% complete of the 1,999 and 2,024 BUSCO genes (Table 7). BUSCO and Compleasm results confirmed the genome assembly’s completeness.
We also aligned NGS short reads and PacBio HiFi long reads to the genome by bwa (v0.7.12) and minimap2 (v 2.24). A total of 99.55% of the reads were mapped, achieving 99.82% genome coverage, which reflects the high quality of the sequencing data. Meanwhile, evaluation of assembly accuracy by Merqury (v1.4.1)57, the quality value (QV) obtained from short-read and long-read alignments were 50.78 and 67.13, both demonstrating further the high quality of assembled genomes.
Data availability
All sequencing data and genome assembly generated in this study have been deposited in public repositories. Raw sequencing data are available in the NCBI under the BioProject accession number PRJNA1273044. Specifically, the Illumina genomic sequencing data (SRR33856795), PacBio HiFi sequencing data (SRR33856794), and Hi-C sequencing data (SRR33856793) are deposited in the NCBI Sequence Read Archive (SRA). The transcriptome Illumina sequencing data from leaves (SRR33856792), barks (SRR33856791), and roots (SRR33856790) are also available in the NCBI SRA under accession number SRP59038152. The final genome assembly has been deposited at NCBI Assembly under accession number GCA_050941835.153. The corresponding GFF annotation file is available at Figshare (https://doi.org/10.6084/m9.figshare.29424683.v1)54.
Code availability
No specific code or script was used in this work. All commands used in the processing were executed according to the manual and protocols of the corresponding bioinformatics software.
References
Fang, J., Guo, Z., Piao, S. & Chen, A. Terrestrial vegetation carbon sinks in China, 1981–2000. Sci. China Ser. D Earth Sci. 50, 1341–1350, https://doi.org/10.1007/s11430-007-0049-1 (2007).
Wu, Z. Y. & Raven, P. H. Flora of China, Vol. 15: Myrsinaceae through Loganiaceae (Science Press, Beijing & Missouri Botanical Garden Press, St. Louis, 1996).
Chen, Y. J., Zhang, H. G. & Li, X. Phenylethanoid glycosides from the bark of Fraxinus mandschurica. Chem. Nat. Compd. 45, 330–332, https://doi.org/10.1007/s10600-009-9349-3 (2009).
Hu, L.-J., Uchiyama, K., Shen, H.-L. & Ide, Y. Multiple-scaled spatial genetic structures of Fraxinus mandshurica Rupr over a riparian-mountain landscape in Northeast China. Conserv. Genet. 11, 77–87, https://doi.org/10.1007/s10592-009-0004-0 (2010).
Liu, J. N. et al. Pan-genome analyses of 11 Fraxinus species provide insights into salt adaptation in ash trees. Plant Commun. 6, 101137, https://doi.org/10.1016/j.xplc.2024.101137 (2025).
Zhang, K. et al. Comparative transcriptome analysis of differentially expressed genes and pathways in male and female flowers of Fraxinus mandshurica. PLOS ONE 19(9), e0308013, https://doi.org/10.1371/journal.pone.0308013 (2024).
He, B. et al. Genome-wide analysis and molecular dissection of the SPL gene family in Fraxinus mandshurica. BMC Plant Biol 22, 451, https://doi.org/10.1186/s12870-022-03838-9 (2022).
Zeng, T. et al. The telomere-to-telomere gap-free reference genome of wild blueberry (Vaccinium duclouxii) provides its high soluble sugar and anthocyanin accumulation. Hortic. Res. 10, uhad209, https://doi.org/10.1093/hr/uhad209 (2023).
J. Liu. NGDC Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/86106/show GWHFDPP00000000.1 (2024).
Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2(2), e107, https://doi.org/10.1002/imt2.107 (2023).
Liu, B. H. et al. Estimation of genomic characteristics by analyzing K-mer frequency in de novo genome projects. Quant. Biol. 35, 62–67, https://doi.org/10.48550/arXiv.1308.2012 (2013).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Cheng, H., Asri, M., Lucas, J., Koren, S. & Li, H. Scalable telomere-to-telomere assembly for diploid and polyploid genomes with double graph. Nat. Methods 21, 967–970, https://doi.org/10.1038/s41592-024-02269-8 (2024).
Rautiainen, M. et al. Telomere-to-telomere assembly of diploid chromosomes with Verkko. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01662-6 (2023).
Antipov, D., et al Verkko2 integrates proximity ligation data with long-read De Bruijn graphs for efficient telomere-to-telomere genome assembly, phasing, and scaffolding. Genome Res. https://doi.org/10.1101/gr.280383.124 (2025).
Peska, V. & Garcia, S. Origin, diversity, and evolution of telomere sequences in plants. Front. Plant Sci. 11, 117, https://doi.org/10.3389/fpls.2020.00117 (2020).
Brown, M. R., Manuel Gonzalez de La Rosa, P. & Blaxter, M. tidk: a toolkit to rapidly identify telomeric repeats from genomic datasets. Bioinformatics 41, btaf049, https://doi.org/10.1093/bioinformatics/btaf049 (2025).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Hortic. Res. 10, uhad127, https://doi.org/10.1093/hr/uhad127 (2023).
Zhang, H. et al. Fast alignment and preprocessing of chromatin profiles with Chromap. Nat. Commun. 12, 6566, https://doi.org/10.1038/s41467-021-26865-w (2021).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39, btac808, https://doi.org/10.1093/bioinformatics/btac808 (2023).
Xu, M. et al. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience 9(9), giaa094, https://doi.org/10.1093/gigascience/giaa094 (2020).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl Acad. Sci. USA 117, 9451–9457, https://www.pnas.org/doi/10.1073/pnas.1921046117 (2020).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275 https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1905-y (2019).
Tempel, S. Using and understanding RepeatMasker. Methods Mol Biol 859, 29–51, https://doi.org/10.1007/978-1-61779-603-6_2 (2012).
Storer, J., Hubley, R., Rosen, J., Wheeler, T. J. & Smit, A. F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mobile DNA 12, 2 https://mobilednajournal.biomedcentral.com/articles/10.1186/s13100-020-00230-y (2021).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Zimin, A. V., Puiu, D., Pertea, M., Yorke, J. A. & Salzberg, S. L. Efficient evidence-based genome annotation with EviAnn. Preprint at bioRxiv 2025.05.07.652745, https://doi.org/10.1101/2025.05.07.652745 (2025).
Sayers, E. W. et al. Database resources of the National Center for Biotechnology Information in 2025. Nucleic Acids Res. 53, D20–D29, https://doi.org/10.1093/nar/gkae979 (2025).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRP513361 (2024).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRP522725 (2024).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRP559292 (2025).
Kim, D. et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 https://academic.oup.com/gigascience/article/10/2/giab008/6137725 (2021).
Huff, M. et al. A high-quality reference genome for Fraxinus pennsylvanica for ash species restoration and research. Molecular ecology resources 22(4), 1284–1302, https://doi.org/10.1111/1755-0998.13545 (2022).
Sayers, E. W. et al. Database resources of the National Center for Biotechnology Information. Nucleic acids research 49(D1), D10–D17, https://doi.org/10.1093/nar/gkaa892 (2021).
Coudert, E. et al. Annotation of biologically relevant ligands in UniProtKB using ChEBI. Bioinformatics (Oxford, England) 39(1), btac793, https://doi.org/10.1093/bioinformatics/btac793 (2023).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368, https://www.nature.com/articles/s41592-021-01101-x (2021).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120, https://doi.org/10.1093/nar/gki442 (2025).
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A. & Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41, e121, https://doi.org/10.1093/nar/gkt263 (2013).
Paysan-Lafosse, T. et al. InterPro in 2022. Nucleic acids research 51(D1), D418–D427, https://doi.org/10.1093/nar/gkac993 (2023).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419, https://doi.org/10.1093/nar/gkaa913 (2021).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics 25(1), 25–29, https://doi.org/10.1038/75556 (2000).
Aramaki, T. et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics (Oxford, England) 36(7), 2251–2252, https://doi.org/10.1093/bioinformatics/btz859 (2020).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research 28(1), 27–30, https://doi.org/10.1093/nar/28.1.27 (2000).
Li, S. F. et al. Chromosome-level genome assembly, annotation and evolutionary analysis of the ornamental plant Asparagus setaceus. Hortic Res 7, 48, https://doi.org/10.1038/s41438-020-0271-y (2020).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J Mol Biol 215, 403–410, https://doi.org/10.1016/s0022-2836(05)80360-2 (1990).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124, https://doi.org/10.1093/nar/gki081 (2005).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRP590381 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_050941835.1 (2025).
Siyang, G. Genome annotation file of the telomere-to-telomere assembly of Fraxinus mandshurica. figshare. Dataset. https://doi.org/10.6084/m9.figshare.29424683.v1 (2025).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Huang, N. & Li, H. compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics 39, btad595, https://doi.org/10.1093/bioinformatics/btad595 (2023).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Acknowledgements
This study was supported by the “Heilongjiang Provincial Natural Science Foundation of China (PL2024A012)”.
Author information
Authors and Affiliations
Contributions
Hongtao Zhao conceived and designed the study; Yanming Zhang and Fang Luo collected the samples; Xiyang Zhao and Qiang Tian performed DNA sequencing and Hi-C experiments; Wa Gao performed RNA sequencing; Qun Cai and Mingwei Tang estimated the genome size; Siyang Guo assembled the genome, and assessed the assembly quality; Dandan Zhang and Zicheng Zhang performed the genome annotation and functional genomic analysis. Dandan Zhang conducted the evolutionary analysis and wrote the initial draft of the manuscript; Siyang Guo revised the manuscript. All authors read, edited, and approved the final manuscript for submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, D., Guo, S., Zhang, Z. et al. Telomere-to-telomere genome assembly of the Fraxinus mandshurica Rupr. Sci Data 12, 1850 (2025). https://doi.org/10.1038/s41597-025-06043-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06043-z
