
Abstract
The evolutionary adaptation of insects to aquatic environments remains a fascinating study area in genomics and evolutionary biology. Here, we present a high-quality chromosome-level reference genome of Donacia provosti (Coleoptera: Chrysomelidae), an ecologically and agriculturally significant aquatic pest species, using a combination of PacBio HiFi, Illunima and Hi-C sequencing technologies. The assembly comprises 15 chromosomes, totaling 1.76 Gb in size, and exhibit high contiguity, with contig and scaffold N50 values of 27 Mb and 71 Mb, respectively. Genome completeness, as assessed by BUSCO, was 98.6%. A total of 1.35 Gb of repeat sequences and 20,130 protein-coding genes were identified in this genome. Moreover, several gene families associated with vision and immune are expanded significantly in this pest. This high-quality D. provosti genome assembly would provide a valuable resource for future investigations into its ecological adaptations and the development of pest control measures.
Similar content being viewed by others


Background & Summary
The water area on Earth accounts for the majority, and many insects have adapted to the aquatic ecosystems. Aquatic insects live in water during one or more phases of their life cycle, such as dragonflies and mayflies, which both lay eggs in water, and only after they have developed and matured do they move onto land1,2. These aquatic insects play important ecological roles as primary consumers, detritivores, predators, and pollinators in both aquatic and terrestrial ecosystems. Aquatic insect groups evolved from terrestrial ancestors through adaptation to freshwater ecosystems3. Freshwater ecosystems exhibit a remarkable diversity of habitats, including ponds, lakes, and ditches. Colonization of these environments required significant evolutionary adaptations in various physiological and behavioral mechanisms, including thermo- and osmoregulation, respiration, and feeding strategies. However, research on insect aquatic adaptation remains limited, the mechanisms driving physiological, morphological, and behavioral changes during their transition to aquatic environments are poorly understood.
The Chrysomelidae, commonly known as “leaf beetles”, comprises approximately 38,000 described species4. The majority of living leaf beetles feed on angiosperms, and many leaf beetles are considered agricultural pests, such as Colorado Potato Beetle (Leptinotarsa decemlineata), western corn rootworm (Diabrotica virgifera), striped flea beetle (Phyllotreta striolata) and coconut leaf beetle (Brontispa longissima). D. provosti (Fairmaire, 1885) (Coleoptera: Chrysomelidae) is a damaging pest of aquatic crops, first recorded in Beijing, China, in 1885. The species distribution includes Russia, South Korea, Japan and China, yet is rapidly spreading globally. In China, it is found from Hainan to Heilongjiang, with significant infestations in Hubei and Jiangsu. D. provosti primarily feeds on lotus and rice, causing notches, holes, and epidermal damage to lotus leaves. Lotus stems and roots are damaged by the larvae, resulting in dark brown spots, decay, and stunted growth, and could make the plant susceptible for fungal infections. In the 2000s, it caused 15%–20% losses in lotus root production in China5. Current control relies on costly chemical pesticides that pollute the water, and pose a threat to human and livestock health. There is an urgent need for environmentally friendly pest management strategies to suppress this pest.
With the advancement of sequencing technologies, their application in entomological research has become increasingly prevalent, and several leaf beetle genomes have now been sequenced. This study presents the first whole-genome sequencing of D. provosti, yielding a high-quality chromosome-level reference genome, and it would provide a valuable resource for future investigations into its ecological adaptations and the development of pest control measures.
Methods
Insect collection and genomic sequencing
Approximately 200 samples of D. provosti were collected from Enshi city(30.25°N,109.05°E), Hubei province, and subjected to a 24-hour laboratory starvation period to minimize contamination of gut content. These samples were subsequently washed with double-distilled water (ddH2O) and ethanol to remove external contaminants, followed by immediate flash-freezing in liquid nitrogen. These samples were then transferred to a −80 °C freezer for long-term storage. Genome sequencing of a female adult was performed using PacBio Revio System with SMRTbell Express Library Prep Kit, generating ~170 Gb HiFi reads and achieving an N50 of ~14 Kb. For Illumina genome sequencing, three short paired-end DNA libraries with a 400-bp insert size were constructed using the TruSeq DNA PCR-Free Library Prep Kit (Illumina) according to the manufacturer’s instructions and sequenced on an Illumina novaseq xplus platform. The total RNA was extracted from three adults, and three short paired-end libraries with a 400-bp insert size was constructed and sequenced on an Illumina novaseq xplus platform. All sequencing work was performed at Berry Genomics Corporation.
Genome assembly and quality assessment
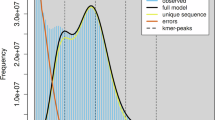
Genome size estimation was conducted through k-mer frequency analysis of PacBio HiFi reads using Jellyfish (v2.1.3)6. The k-mer counting process was executed using the following Jellyfish command: jellyfish count -m 17 -C -s 100 M -t 20, which revealed an estimated genome size of 1.7 Gb.
The PacBio reads were assembled using Hifiasm (ve0.20.0-r639)7 with parameters: -l 3, generating an initial assembly comprising 4,990 contigs with a total length of approximately 2.2 Gb and a contig N50 of 21 Mb. To eliminate redundant sequences, the assembly was further processed using purge_dups (v1.2.3)8 with parameters set to -2 -a 50. This purification step yielded a refined primary assembly with a total length of 1.76 Gb and an improved contig N50 of 27 Mb (Table 1).
Genome scaffolding
We sequenced an adult sample, generating approximately 150 Gb of Hi-C paired-end reads. Raw reads underwent quality control using fastp (v0.23.1)9, resulting in a Q30 score of 92.3%. Subsequently, these reads were aligned to the reference genome using Bowtie2 (v2.4.1)10. Valid chromatin interactions were identified and filtered (removing multiple hits and singletons) using HiC-Pro (v2.11.0)11. Finally, contigs were anchored and oriented into 15 scaffolds (Fig. 1a,b) with YAHS (v1.2.2)12, which correspond to the 15 actual chromosomes13. This chromosome-level genome assembly achieved a scaffold N50 of 71 Mb, with the longest contig measuring 127 Mb and the shortest at 55 Mb. The circos plot was drawn by TBtools (v2.326)14.

Heatmap of genome-wide Hi-C data and circular representation of the chromosomes of D. provosti. (a) The heatmap of chromosome interactions in D. provosti, with densities calculated in 500 Kb windows. The frequency of Hi-C interaction links is represented by colours, which ranges from yellow (low) to red (high); (b) Circular representation of the chromosomes. Two tracks represent the distribution of gene density (line plot), TE density (bar plot), with densities calculated in 10 Kb windows, and TEs with overlaps in the RepeatMasker results were removed. Both gene density and TE density are ranging from 0 to 100%.
Genome annotation
The annotation of Transposable Elements(TEs) was performed through a comprehensive repeat analysis pipeline. Initially, a de novo repeat library was constructed using RepeatModeler (v2.0.1)15 with the NCBI BLAST engine as the search algorithm (-engine ncbi). Subsequently, RepeatMasker (v4.0.5)16 was employed to identify TEs by integrating both the de novo repeat library and TE databases (RepBase 20170127). Finally, A total of 1.35 Gb (76.6% of the 1.76 Gb genome) repeat sequence was identified (Table 1). While automated annotation methods can be limited in their precision, particularly for genomes with high TEs densities, and coupled with the lack of curated insect TEs databases, this can introduce biases in TEs annotation.
To ab initio predict coding genes, we utilized repeat-masked genome sequences with AUGUSTUS (v2.7)17. For homology-based prediction, protein sequences of Chrysomelidae species were retrieved from the NCBI and UniProt databases. These sequences were subsequently mapped to the genome using exonerate (v2.4.0)18, and incomplete gene models which lacking both start and stop codons were filtered out and removed. Quality-controlled reads from RNA libraries were mapped to the genome using Bowtie2 (v2.4.1)10. StringTie (v2.1.1)19 was then employed to construct gene prediction models based on these alignments. Finally, the gene predictions generated through these three approaches were integrated using the EVidenceModeler (v1.1.1)20. The transcriptomic evidence was weighted by a factor of 10, ab initio evidence by a factor of 4, and homologous annotation evidence by a factor of 1. A total of 20,130 protein-coding gene models were predicted (Table 1). Functional annotation was performed by aligning all genes to these four databases, NCBI-NR, KEGG, InterPro, and GO database with diamond (v0.9.19)21 and interproscan (v5.1)22.
Phylogenetic analysis
Five additional Chrysomelidae species (Table 2), Acanthoscelides obtectus, Diabrotica virgifera, Diorhabda carinulata, Phaedon cochleariae, Phyllotreta striolata were used to infer orthologous genes, and Tribolium castaneum was select as outgroup. Genome assembly and annotation of these five Chrysomelidae species A. obtectus23, D. virgifera24, D. carinulata25, P. cochleariae26, P. striolata27, and T. castaneum28 was download from NCBI genome database. Orthologous gene clusters were identified using the OrthoFinder algorithm. Subsequently, species phylogenetic tree were constructed based on conserved single-copy gene sequences using FastTree (v2.1.10)29 with the JTT + CAT model. Divergence times were then estimated, and gene family expansion and contraction were analyzed. All gene family-related analyses and visualizations were further facilitated by OrthoVenn330. The calibration times for divergence estimation were set according to data from TimeTree31, with a minimum of 152 million years ago (MYA) and a maximum of 236 MYA for T. castaneum and A. obtecta. The results revealed that D. provosti diverged from its closest relative, A. obtectus, approximately 0.6 MYA, and T. castaneum around 2.3 MYA (Fig. 2a). Through the analysis of gene family expansion and contraction, we identified 38 expanded gene families and 406 contracted gene families in D. provosti. GO enrichment analysis of the expanded gene families, which conducted utilizing the OmicShare tools32, revealed significant enrichment in vision-related and immune-related functions, such as response to blue light (GO:0009637) and mannan binding (GO:2001065) (Fig. 2b). GO enrichment analysis for the contracted gene families showed that only one GO term was found to be significantly enriched (GO:0003008, system process).

Phylogenetic analysis. (a) Phylogeny and orthology analyses between D. provosti and other Chrysomelidae species, and T. castaneum was select as outgroup. The expanded (Red) and Contracted (Blue) gene families are presented alongside the species and nodes; (b) GO enrichment of expand gene family of D. provosti.
Data Records
Raw sequence reads and genome assembly are available on NCBI Sequence Read Archive database with accession numbers BioProject PRJNA123808333; Pacbio, Hi-C, Illumina and transcriptome sequencing reads have been deposited in the Sequence Read Archive (SRA) databases with the accession number of SRP57196134; Genome assembly has been deposited at the NCBI under the accession number of JBMHBG00000000035; datasets include genome assembly and gene annotation are deposited on figshare36.
Technical Validation
The accuracy and completeness of genome assembly and gene annotation were validated using a multi-approach. First, Illumina reads were quality-filtered using fastp (v0.23.1)9 with Q30 = 93.4%, then quality-filtered reads mapped to the assembled contigs using BWA (v0.7.5a)37, and the resulting alignment processed via SAMtools (v1.19.2)38 revealed a mapping rate of 98.90%, and a coverage rate of 97.9%. Second, RNA-Seq reads from three whole-body transcriptomes were quality-filtered using fastp (v0.23.1)9 with Q30 = 93–95%, and alimented to the genome assembly with hiast2 (v2.2.1)39, the results indicated that >95% of reads aligned to coding regions. Third, BUSCO (v5.2.2)40 analysis for the genome assembly using the Insecta odb10 dataset (-l insecta_odb10 -m genome) identified 98.6% of expected insect single-copy orthologs, with 98.6% were classified as complete, comprising 1.8% duplicated genes, while 0.4% were fragmented. To further ensure the comprehensiveness of the gene annotation, BUSCO (v5.2.2)40 was also employed with the Insecta odb10 database (-l insecta_odb10 -m prot), identifying 97.2% of conserved single-copy orthologs in the annotated protein set (97.2% complete, 1.7% duplicated, and 0.4% fragmented). RNA-Seq analysis of three whole-body transcriptomes demonstrated expression of 18,320 (87%) annotated genes in at least one sample. Finally, homology searches against NCBI-NR, KEGG, InterPro, or GO databases revealed significant sequence similarity for 19,635 (98%) of predicted gene models in at least one database.
Data availability
The dataset is available at NCBI Sequence Read Archive database with accession numbers BioProject PRJNA1238083; Raw sequencing reads have been deposited in the Sequence Read Archive (SRA) databases with the accession number of SRP571961; Genome assembly has been deposited at the NCBI under the accession number of JBMHBG000000000.
Code availability
No specific code or script was used in this work. Commands used for data processing were all executed according to the manuals and protocols of the corresponding software.
References
Stoks, R. & Córdoba-Aguilar, A. Evolutionary Ecology of Odonata: A Complex Life Cycle Perspective. Annual Review of Entomology 57, 249–265, https://doi.org/10.1146/annurev-ento-120710-100557 (2012).
Jacobus, L. M., Macadam, C. R. & Sartori, M. Mayflies (Ephemeroptera) and Their Contributions to Ecosystem Services. Insects 10, 170, https://doi.org/10.3390/insects10060170 (2019).
Wootton, R. J. The historical ecology of aquatic insects: An overview. Palaeogeography, Palaeoclimatology, Palaeoecology 62, 477–492, https://doi.org/10.1016/0031-0182(88)90068-5 (1988).
Wilf, P. et al. Timing the Radiations of Leaf Beetles: Hispines on Gingers from Latest Cretaceous to Recent. Science 289, 291–294, https://doi.org/10.1126/science.289.5477.291 (2000).
Zhan, H. et al. Molecular Characterization of Donacia provosti (Coleoptera: Chrysomelidae) Larval Transcriptome by De Novo Assembly to Discover Genes Associated with Underwater Environmental Adaptations. Insects 12, 281, https://doi.org/10.3390/INSECTS12040281 (2021).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359, https://doi.org/10.1038/nmeth.1923 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology 16, 259, https://doi.org/10.1186/s13059-015-0831-x (2015).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39, https://doi.org/10.1093/bioinformatics/btac808 (2022).
Petitpierre, E. in Biology of Chrysomelidae (eds Jolivet, P., Petitpierre, E. & Hsiao, T. H.) 131–159 (Springer Netherlands, 1988), https://doi.org/10.1007/978-94-009-3105-3_9 (1988).
Chen, C. et al. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Molecular plant 13, 1194–1202, https://doi.org/10.1016/j.molp.2020.06.009 (2020).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr Protoc Bioinformatics 25, 4.10.11–14.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–W439, https://doi.org/10.1093/nar/gkl200 (2006).
Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31, https://doi.org/10.1186/1471-2105-6-31 (2005).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12, 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
Blum, M. et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res 49, D344–D354, https://doi.org/10.1093/nar/gkaa977 (2021).
NCBI GenBank https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_963669975.1/ (2023).
NCBI GenBank https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_917563875.1/ (2022).
NCBI GenBank https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_026250575.1/ (2022).
NCBI GenBank https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_918026855.4/ (2022).
NCBI GenBank https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_918026865.1/ (2022).
NCBI GenBank https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_031307605.1/ (2023).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Molecular Biology and Evolution 26, 1641–1650, https://doi.org/10.1093/molbev/msp077 (2009).
Sun, J. et al. OrthoVenn3: an integrated platform for exploring and visualizing orthologous data across genomes. Nucleic Acids Res 51, W397–W403, https://doi.org/10.1093/nar/gkad313 (2023).
Kumar, S. et al. TimeTree 5: An Expanded Resource for Species Divergence Times. Molecular Biology and Evolution 39, https://doi.org/10.1093/molbev/msac174 (2022).
Mu, H. et al. OmicShare tools: A zero-code interactive online platform for biological data analysis and visualization. iMeta 3, e228, https://doi.org/10.1002/imt2.228 (2024).
NCBI BioProject https://identifiers.org/ncbi/bioproject:PRJNA1238083 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP571961 (2025).
NCBI Genbank https://identifiers.org/ncbi/insdc.gca:GCA_052550795.1 (2025).
Wang, H. The genome and annotation of Donacia provosti. figshare. Dataset. https://doi.org/10.6084/m9.figshare.28639664.v2 (2025).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Acknowledgements
This work was supported by China Agriculture Research System of MOF and MARA 359 (CARS-24-C-03).
Author information
Authors and Affiliations
Contributions
H.L. and C.L. designed the research; H.L. performed the research and improved by L.M., Y.X., C.W. and C.G.; H.L. wrote the paper, which was improved by M.H.A.; L.L. provide suggestions and article revision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, H., Ma, L., Sun, Y. et al. Chromosome-Level Genome Assembly of Donacia provosti. Sci Data 12, 1765 (2025). https://doi.org/10.1038/s41597-025-06051-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06051-z
