
Abstract
The Japanese cutlassfish (Trichiurus japonicus) is a commercially important marine species across Asia. Here, we present a high-quality, chromosome-level genome assembly generated using PacBio HiFi, Hi-C, and Nanopore ONT reads. The nuclear genome comprised 24 chromosomes with 160 scaffolds totaling 1,138 Mb, with a scaffold N50 of 47.10 Mb and an average scaffold length of 6.18 Mb. A complete mitochondrial genome of 16,796 bp was also assembled, comprising 13 protein-coding and 23 non-coding RNA (ncRNA) genes, with 99.32% sequence identity to the reference in the NCBI database. The nuclear genome encodes 26,541 protein-coding genes (median length: 7,391 base pairs) and 16,383 non-coding RNA (ncRNA) genes. The ncRNA genes account for approximately 0.1694% of the genome’s total length. BUSCO analysis indicated 99.4% and 99.2% completeness against the Actinopterygii ortholog set for the genome and proteome. Functional annotation covered 98.15% of genes. Recognized repeat elements and ncRNA regions accounted for 61.10% of the nuclear genome. With high mapping rates from external datasets, this assembly offers a valuable foundation for future sequencing-based studies.
Similar content being viewed by others
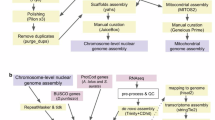

Background & Summary
The Japanese cutlassfish, Trichiurus japonicus, also known as large-head hairtail, is widely distributed in the northwestern Pacific Ocean, inhabiting coastal waters at depths of 100–200 meters and exhibiting diurnal vertical migration1. It is a commercially important species in East Asia’s capture fisheries and the dominant Trichiuridae species in most fishing ports across Taiwan, accounting for 40% to 100% of reported catches2. The global capture production of Trichiuridae, which often aggregates the output of T. japonicus under the globally distributed species T. lepturus as it was previously regarded as a subspecies, has exceeded one million tons annually since 19943, with over 80% caught from the East Asian Seas. Compared to other Trichiuridae species, the broad latitudinal distribution of the Japanese cutlassfish suggests that this temperate species is well adapted to cold waters4, leading to the hypothesis of temperature-driven speciation5.
A decline in the Japanese cutlassfish population has been documented in the literature1, putatively caused by overfishing and the impacts of global climate change. To assess the stock structure for resource monitoring, it is crucial to use a methodology that can accurately assign species rather than relying solely on phenotypic features. Using molecular features such as the mitochondrial control region, cytochrome b (cyt b), or cytochrome c oxidase I (COI), researchers successfully assessed the cutlassfish population composition2,3. In light of the heavy exploitation of Japanese cutlassfish, developing sophisticated genome-derived molecular markers to elucidate genetic diversity within populations is critical for informing resource management strategies.
The other genome of T. japonicus (Genebank assembly: GCA_046254865.1) was published by Shanghai Ocean University on the NCBI database in Dec 2024. This genome assembly, consisting of 24 chromosomes (931.81 Mb) and 1,014 unplaced scaffolds with a scaffold N50 of 39.2 Mb, was constructed by integrating Oxford Nanopore long reads (112 Gb), Illumina short reads (66.37 Gb), and Hi-C scaffolding data (104.11 Gb). This manuscript presents a more complete genome assembly of T. japonicus, built from PacBio HiFi Reads, Oxford Nanopore, 10x, Omni-C, and Hi-C data. To maximize the utility of read information from different sequencing platforms, we combined the assembling processes of HiFiasm6,7, and SALSA28, and further enhanced genome continuity using our gap-filling algorithm, LOCLA9. The final genome assembly comprises 24 chromosomes and 160 scaffolds, totaling 1,138 Mb in length, with a scaffold N50 length of 47.10 Mb, an average scaffold length of 6.18 Mb, and a complete mitogenome of 16,796 bp. Comprehensive annotation was performed on the nuclear and mitochondrial genomes to identify protein-coding and non-coding genes. This assembly will provide a foundational infrastructure for further study.
Methods
Sample collection and library construction
A female Japanese cutlassfish (Trichiurus japonicus), measuring approximately 58 cm in length and weighing 117 grams, was captured offshore of Kaohsiung, Taiwan (22°15.754′ N, 120°38.784′ E) in May 2020. Tissue samples from the brain, heart, blood, spleen, kidney, liver, and muscle were collected on board the fishing vessel. High-quality genomic DNA was extracted from various tissues using the DNeasy Blood & Tissue Kit (Qiagen, USA) following the manufacturer’s instructions. DNA quality and concentration were assessed using a NanoDrop One spectrophotometer (Thermo Scientific, USA) and an automated pulsed-field capillary electrophoresis system (Agilent, USA). Liver-derived genomic DNA demonstrated the highest quality among all tissue sources and was used for library preparation for short-read, long-read, and Hi-C sequencing platforms.
Genome sequencing
A single-molecule real-time (SMRT) library prepared from extracted liver genomic DNA (gDNA) was sequenced on the PacBio Sequel II platform using an 8 M SMRT Cell chip with a 30-hour movie time. HiFi reads (Q > 20) were generated from circular consensus (CCS) reads, processed with SMRT Link (version: 11.1.0.166339). Two PacBio HiFi cells generated 922,338 and 910,177 reads, respectively, producing 16,520,702,091 bp (average read length: 17,911 bp) and 16,053,456,774 bp (average read length: 17,637 bp), with an average base quality score of 30.
A liver-derived genomic DNA library was constructed and sequenced for Nanopore long-read sequencing using the Oxford Nanopore Technologies (ONT) R9.4 flow cell. Reads longer than 10 kb were retained for subsequent genome assembly. A total of 532,138 reads were generated, yielding approximately 12 Gb of sequence data, with an average read length of 22,575 bp.
The experiment design included high-throughput chromosome conformation capture (Hi-C) sequencing for scaffolding. Hi-C libraries were prepared using chromatin crosslinking protocols to preserve long-range genomic interactions. Sequencing on the Illumina Hi-Seq. 2500 platform generated 42,628 megabase pairs (Mbp) of 151 bp paired-end reads.
Genome assembly and quality evaluation
A contig-level draft genome, based on PacBio HiFi and Hi-C reads, was constructed using HiFiasm (version: 0.19.5-r587)6,7 and is summarized in Table 1. It comprises 1,010 scaffolds with a maximum scaffold length of 28,308,023 bp a contig N50 of 7,895,182 bp. A two-step scaffolding strategy was employed to connect contigs approaching chromosome-level assembly. First, Hi-C reads were reused for scaffolding with SALSA2 (docker version 2.3)8. Subsequently, the global-contig-based (GCB) scaffolding module of LOCLA9 was applied to the SALSA2-derived scaffolds with ONT long reads (over 10 Kbp) to improve scaffold continuity further. After the scaffolding process, 184 scaffolds were assembled, with the maximum scaffold length increased to 55,973,360 bp. Both the contig N50 and mean scaffold length were elevated to 46,998,866 bp and 6,168,641 bp, respectively (Table 1). We applied the LOCLA global-contig-based (GCB) gap-filling module in the last gap-filling step with PacBio long-read data. Among the final 184 scaffolds, 29 were longer than 1 Mb, comprising 1,138,003,112 bp with non-N bases ratio of 0.02%. Meanwhile, the complete mitochondrial genome with a length of 16,796 bp was identified and reconstructed with PacBio
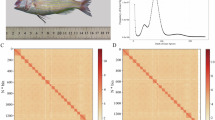
HiFi reads and Canu. The Hi-C contact map (Fig. 1) reveals strong interactions within chromosomes. The final assembly was then further scanned with the FCS-Adapter and FCS-GX (the database was constructed on March 27, 2024). No adapters or contamination were found in the representative nuclear and mitochondrial genomes. In addition, analysis of HiFi reads and final assembly with Merqury (version: 1.3)10 revealed that the average quality value (QV) was 38.59, and k-mer completeness was 86.82%.

Hi-C interaction heatmap for T. japonicus final genome assembly. The resolution for resolving interactions was 2 Mbp, and the data were normalized by read coverage. The grid spacing in the plot was 50 Mbp. The highlighted block represented 24 chromosomes of the T. japonicus nuclear genome.
Repeat element annotation
Repeat elements were annotated through homology-based and de novo prediction approaches. A combined homology database was established using partitions 0, 2, and 6 of Dfam 3.8 (Dfam TE Tools Container v1.89.2)11 and Repbase (2018/10/26)12, limited to the taxonomic level of Actinopterygii. TRF (v4.09)13 and RepeatModeler (v2.0.5)11, which in turn utilize RepeatScout (v1.0.6)12, RECON (v1.08)14, and LTR_retriever (v2.9.0)15, were used for de novo repeat discovery. In total, 4,672,592 elements were identified, spanning 695,266,837 bp and covering 61.10% of the genome (Table 2). These included Class II DNA transposition at 20.77%, long terminal repeats (LTRs) at 9.00%, long interspersed nuclear elements (LINEs) at 8.36%, simple repeat at 2.95%, rolling circle transposons (RC) at 1.77%, short interspersed nuclear elements (SINEs) at 0.87%, satellites at 0.80%. Notably, repeat patterns that did not match any known transposable elements (classified as “Unknown”) occupied 15.67% of the genome (Fig. 2).

The genome annotation of the Japanese cutlassfish. The genomic features of Trichiurus japonicus were summarized using a Circos plot comprising 24 chromosomes. From the outermost to the innermost track, the plot displayed the density of protein-coding genes, Class II DNA transposons, SINEs, LTRs, LINEs, and GC content, calculated in 0.1 Mb windows.
Annotation on protein-coding and non-coding RNA genes
Braker316 annotation pipeline (docker image: braker3: v3.0.7.6) was applied to the genome from the previous section after additional TRF soft-masking for gene structure prediction17,18,19,20, with both transcriptional data and protein alignments as evidence. RNA-seq data were downloaded from the NCBI SRA, including SRR32402605 (adult blood), SRR32402606 (adult muscle), SRR32402607 (adult kidney), SRR32402608 (adult swim bladder), SRR32402609 (adult skin), SRR32402610 (adult eye), SRR32402611 (adult gill), SRR32402612 (adult brain), SRR32402614 (adult liver), and SRR32402615 (adult heart). Raw reads, totaling 64.3 Gbp, were preprocessed with Cutadapt (v4.9)21 to remove library adapters, reads shorter than five bp, and low-quality bases at the 3′-end, and were aligned to the Japanese cutlassfish reference genome using STAR (v 2.7.11b)22. Properly mapped read pairs were retained using samtools (version 1.21)23. Protein sequences from related teleost species were compiled, including proteins in the OrthoDB (version 12, https://bioinf.uni-greifswald.de/bioinf/partitioned_odb12/), Actinopterygii (Taxon ID: 7898) proteins from UniProtKB24, and Scombriformes (Taxonomy ID: 1489894) proteins from the annotated typical genome in NCBI, as evidence for protein-coding gene prediction. Sequence preparation followed Braker3 team’s instructions16. Furthermore, BUSCO lineage “actinopterygii_odb10” was used to help reduce the error rate. For species with multiple full genome assemblies available in NCBI, the primary assembly was used for protein sequence extraction. As a result, 26,541 protein-coding genes comprising 488,952 exons were annotated with a median length of 7,391 (Table 3), among which 39,048 mRNAs were derived, and 2,233 genes were single-exon genes.
The unmasked nuclear genome assembly was used as input for predicting non-coding RNA (ncRNA) genes. The tRNA genes and pseudogenes were identified using tRNAscan-SE (v2.0.12) with a two-step filtering process. First, tRNAscan-SE was applied with parameters -E -I --detail -H to detect all potential tRNA sequences. Second, the EukHighConfidenceFilter was applied with default cutoff settings to distinguish high-confidence tRNAs from pseudogenes. The EukHighConfidenceFilter default criteria were: (i) secondary filtering domain-specific model score ≥50, (ii) secondary filtering secondary structure score ≥10, (iii) secondary filtering isotype-specific model score ≥70, (iv) tertiary filtering isotype-specific model starting score ≥70, and (v) tertiary filtering maximum isotype-specific model score ≤95. Sequences not meeting these criteria were classified as pseudogenes. Infernal (v1.1.5, parameter: -Z 2276.006224 --cut_ga --rfam –nohmmonly --fmt 2 --clanin Rfam.clanin --oskip Rfam.cm)25, referring to the Rfam database (v15.0)26 was used to predict other ncRNA types including, rRNAs, snRNAs, snoRNAs, miRNA precursors, and signal recognition particle RNAs. This approach detects ncRNA families with well-defined primary sequence and conserved secondary structure motifs. Because most lncRNAs lack conserved secondary structures and are not comprehensively represented in Rfam, only a small number of known lncRNA families were annotated in this study. A more complete lncRNA annotation will require full-length transcriptome sequencing and specialized discovery pipelines in future work.
A total of 16,383 ncRNA genes were predicted, including 12,075 tRNAs, 1,728 rRNAs, 1,554 snRNAs, 849 miRNAs, 40 Metazoan signal recognition particle RNA (Metazoa SRP), 113 MALAT1-associated small cytoplasmic RNAs/MEN beta RNAs (mascRNA-menRNA), and other minor ncRNA types, accounting for 0.1694% of the genome. Additionally, 6,699 pseudogenes and 244 cis-regulatory elements (cis reg elements) were predicted in tRNAscan-SE (Table 4).
The mitogenome of T. japonicus was annotated using MITOS2 (v2.1.9), setting parameter: --best, applying the vertebrate mitochondrial genetic code (code 2) and referencing RefSeq. 89 Metazoa dataset. As a result, 13 protein-coding genes, 21 tRNA genes, and 2 rRNA genes were identified (Fig. 3, Table 5). The annotation plot of the mitochondrial genome was generated with OGDRAW (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html).

Circular map of the Trichiurus japonicus mitochondrial genome (16,796 bp). Protein-coding genes are color-coded by respiratory complex: Complex I (yellow), Complex IV (pink), ATP synthase (green), and other genes (purple). Transfer RNAs (blue) and ribosomal RNAs (red) were also shown here. The inner grey ring represented the GC content calculated in 4-bp sliding windows.
Functional annotation of proteins
Methods for annotating functions and structural domains of putative proteins from Japanese cutlassfish genome were applied to both nuclear and mitochondrial proteins, comprising a total of 39,061 proteins from 26,554 genes: DIAMOND (v2.1.10.164, parameters: --header simple --max-target-seqs. 1 --outfmt 6 qseqid stitle pident length mismatch gapopen qstart qend sstart send evalue bitscore staxids --evalue 0.001)27 for sequence homology searches against the NCBI Non-redundant protein database28; KEGG pathway assignment using KEGG Automatic Annotation Server (KAAS, v2.1, model: ghostz model with BBH method)29,30 against 38 fish species comprising 1,034,578 sequences; InterProScan (v5.73–104.0, paremeters: -goterms)31,32 for protein domain detection; and DeepTMHMM (v1.0.24)33 accompanied by the DeepTMHMM_parser (Github: https://github.com/soldatsm/ DeepTMHMM_parser) for transmembrane feature detection. Gene Ontology was derived from InterProScan results.
The DIAMOND blastp results showed that 98.70% of the protein sequences, corresponding to 98.15% of the genes (Table 6), had homologs in the nr database. Of these identified proteins, 88.54% were informative, originating from 85.39% of the total genes. The top three species most frequently assigned by DIAMOND blastp were Thunnus maccoyii, T. albacares, and T. thynnus, accounting for 50.70% of all annotated proteins (Table 7).
The performance of annotations from these tools was visualized using a Venn diagram generated with the R package Venn (GitHub: https://github.com/dusadrian/venn). All applied tools annotated proteins from 17,700 genes (66.69% of all genes), whereas 1,506 genes (5.67%) received no annotation (Fig. 4).

Venn diagram illustrating the annotation coverage across different tools. The pink area with a red border represented all protein-coding genes (“all”, for nuclear + mitochondrial, 26,554 genes). More than half of the genes were annotated by all applied methods, whereas 1,506 genes (5.67%) lacked any annotation information.
DeepTMHMM analysis detected 5,746 (14.72%) alpha-helical transmembrane proteins without a signal peptide (TM), 2,764 (7.08%) alpha-helical transmembrane proteins with a signal peptide (SP + TM), 3,192 (8.17%) globular proteins with a signal peptide (SP), 27,346 (70.03%) globular proteins without a signal peptide (GLOB), and 13 (0.03%) beta-barrel transmembrane proteins (BETA) (Table 8).
Data Records
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JBDZUI000000000. The version described in this paper is JBDZUI010000000. The associated raw sequence data are under accession SRP58275134 and the assembly under accession GCA_050140585.135. The raw reads of PacBio HiFi (SRR33408711), Nanopore (SRR33408712), 10x (SRR33408710), and Hi-C (SRR33408713) can be retrieved under this SRP archive. Genome annotation files (GFF3, GTF and FASTA formats), repeat sequence annotations, and the complete mitochondrial genome assembly are available through Zenodo36. All datasets are publicly accessible and adhere to open-access data sharing principles, ensuring reproducibility and enabling future research applications.
Technical Validation
Genome completeness of the assembly built in this study was assessed using BUSCO (version: 5.8.2)37 with the actinopterygii_odb10 reference dataset, which comprised 3,640 conserved single-copy orthologs (BUSCOs) derived from 26 teleost genomes. The genome recovered 3,617 (99.4%) markers completely, including 2,925 (80.4%) complete single-copy BUSCOs and 692 (19.0%) complete and duplicated BUSCOs (Table 9). Fragmented and missing BUSCOs were 15 (0.4%) and 8 (0.2%), respectively. For the predicted proteome, 3,612 (99.2%) BUSCOs were recovered as complete BUSCOs; fragmented and missing BUSCOs were 10 (0.3%) and 18 (0.5%), respectively.
The assembled mitochondrial genome was compared to the published complete T. japonicus mitochondrial genome available on NCBI accession: NC_011719.1 using the BLASTn algorithm, revealing a sequence similarity of 99.32%. To assess the suitability of the assembly as a representative genome for sequence-based study, two raw read datasets from NCBI SRA data repository, originally for a genome assembling pipeline, were utilized to evaluate mapping efficacy: SRR32395546, a HiSeq read set in 150 bp pair-end format generated on Illumina platform, and SRR32395604, a long-read dataset of Oxford Nanopore Technologies. The Illumina short reads were preprocessed with Cutadapt (v4.9)21 (parameters: -cut 60 --cut -60 --discard --error-rate 0.25 --overlap 10 -m 500 --max-aer 0.15), and the ONT reads were preprocessed with Porechop (v0.2.4, https://github.com/rrwick/Porechop). Both read sets’ mapping rates and other indicators were accessed using QUAST (v5.2.0, parameter: -large)38 (Table 10). The mapping rates of short and long reads were 99.39% and 99.94%, respectively, indicating that most reads could be successfully aligned to the genome assembly presented in this study. The quartet (v1.2.5)39 TeloExplorer and CentroMiner modules were applied to identify telomere and centromere (Fig. 5). Motifs for telomere and centromere detection were based on species-specific or known consensus sequences. Evidence from transposable elements (TE) were used to assist in centromere determining, as generated by EDTA (version: 2.2.2)40. TEs overlapping with coding DNA sequences (CDS) predicted by BRAKER3 were excluded from the analysis.

The recognized telomere and centromere inside the Japanese cutlass genome.
Data availability
The whole-genome shotgun assembly has been deposited at DDBJ/ENA/GenBank under accession JBDZUI000000000 (version JBDZUI010000000). Raw sequencing reads are available in the NCBI SRA under BioProject SRP582751 (PacBio HiFi: SRR33408711; Nanopore: SRR33408712; 10x Genomics: SRR33408710; Hi-C: SRR33408713). The genome assembly (GCA_050140585.1), annotation files (GFF3, GTF, FASTA), repeat sequence annotations, and the complete mitochondrial genome are accessible through Zenodo (https://zenodo.org/records/15686396). All datasets are publicly available without restriction.
Code availability
All scripts and commands followed the instructions in GitHub (https://github.com/Abieskawa/Annotation-Toolkit) for each bioinformatic tool. The LOCLA used to polish the assembly is available at https://github.com/lsbnb/locla.
References
The Fish Database of Taiwan. (Academia Sinica Center for Digital Cultures, 2015).
Wang, H.-Y., Dong, C. A. & Lin, H.-C. DNA barcoding of fisheries catch to reveal composition and distribution of cutlassfishes along the Taiwan coast. Fisheries Research 187, 103–109, https://doi.org/10.1016/j.fishres.2016.11.015 (2017).
Food and Agriculture Organization of the United Nations. Fisheries Department. (Food and Agriculture Organization of the United Nations, Rome, 2024).
Lin, H.-C., Tsai, C.-J. & Wang, H.-Y. Variation in global distribution, population structures, and demographic history for four Trichiurus cutlassfishes. PeerJ 9, https://doi.org/10.7717/peerj.12639 (2021).
Tzeng, C. H., Chen, C. S. & Chiu, T. S. Analysis of morphometry and mitochondrial DNA sequences from two Trichiurus species in waters of the western North Pacific: taxonomic assessment and population structure. Journal of Fish Biology 70, 165–176, https://doi.org/10.1111/j.1095-8649.2007.01368.x (2007).
Cheng, H. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nature Biotechnology 40, 1332–1335, https://doi.org/10.1038/s41587-022-01261-x (2022).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Ghurye, J. et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLOS Computational Biology 15, e1007273, https://doi.org/10.1371/journal.pcbi.1007273 (2019).
Chuang, W.-H. et al. A Novel Genome Optimization Tool for Chromosome-Level Assembly across Diverse Sequencing Techniques. bioRxiv, 2023.2007.2020.549842, https://doi.org/10.1101/2023.07.20.549842 (2023).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), i351–358, https://doi.org/10.1093/bioinformatics/bti1018 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res 12, 1269–1276, https://doi.org/10.1101/gr.88502 (2002).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Research 46, e126–e126, https://doi.org/10.1093/nar/gky730 (2018).
Bruna, T., Gabriel, L. & Hoff, K. Navigating Eukaryotic Genome Annotation Pipelines: A Route Map to BRAKER, Galba, and TSEBRA. (2024).
Gabriel, L. et al. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. bioRxiv, https://doi.org/10.1101/2023.06.10.544449 (2024).
Brůna, T., Lomsadze, A. & Borodovsky, M. GeneMark-ETP significantly improves the accuracy of automatic annotation of large eukaryotic genomes. Genome Res 34, 757–768, https://doi.org/10.1101/gr.278373.123 (2024).
Pertea, G. & Pertea, M. GFF Utilities: GffRead and GffCompare [version 2; peer review: 3 approved]. F1000Research 9, https://doi.org/10.12688/f1000research.23297.2 (2020).
Quinlan, A. R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr Protoc Bioinformatics 47, 11.12.11–34, https://doi.org/10.1002/0471250953.bi1112s47 (2014).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17, 3, 10.14806/ej.17.1.200 (2011).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, https://doi.org/10.1093/bioinformatics/bts635 (2013).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Coudert, E. et al. Annotation of biologically relevant ligands in UniProtKB using ChEBI. Bioinformatics 39, btac793, https://doi.org/10.1093/bioinformatics/btac793 (2023).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Ontiveros-Palacios, N. et al. Rfam 15: RNA families database in 2025. Nucleic Acids Research 53, D258–D267, https://doi.org/10.1093/nar/gkae1023 (2025).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nature Methods 18, 366–368, https://doi.org/10.1038/s41592-021-01101-x (2021).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35, D61–65, https://doi.org/10.1093/nar/gkl842 (2007).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35, W182–185, https://doi.org/10.1093/nar/gkm321 (2007).
Suzuki, S., Kakuta, M., Ishida, T. & Akiyama, Y. Faster sequence homology searches by clustering subsequences. Bioinformatics 31, 1183–1190, https://doi.org/10.1093/bioinformatics/btu780 (2015).
Blum, M. et al. InterPro: the protein sequence classification resource in 2025. Nucleic Acids Research 53, D444–D456, https://doi.org/10.1093/nar/gkae1082 (2025).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
Hallgren, J. et al. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv https://doi.org/10.1101/2022.04.08.487609 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP582751 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_050140585.1 (2025).
Lab of Systems and Network Biology et al. Tachiuo. Zenodo. https://doi.org/10.5281/zenodo.15686396 (2025).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molecular Biology and Evolution 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075, https://doi.org/10.1093/bioinformatics/btt086 (2013).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Horticulture Research 10, https://doi.org/10.1093/hr/uhad127 (2023).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biology 20, 275, https://doi.org/10.1186/s13059-019-1905-y (2019).
Acknowledgements
This research was funded by the Ministry of Agriculture, Taiwan (Project No. 109AS-1.2.2-FA-F4, awarded to CLAC and CYL), and the National Science and Technology Council, Taiwan (Grants NSTC 110-2320-B-038-087 and NSTC 113-2221-E-038-020-MY3, awarded to SHC). We sincerely thank the Taiwan Ocean Conservation and Fisheries Sustainability Foundation and Ms. Shu-Jen Ho for their assistance in obtaining the beltfish samples. We also thank Ms. Jeng-Yi Li and the High Throughput Sequencing Core in the Biodiversity Research Center at Academia Sinica for performing the PacBio HiFi sequencing. The core facility is funded by the Academia Sinica Core Facility and Innovative Instrument Project (AS-CFII-108-114).
Author information
Authors and Affiliations
Contributions
C.Y.L. and S.H.C. conceived the study and provided overall supervision. C.L.A.C. initiated the project and was responsible for sample collection, library preparation, and sequencing. P.C.H., C.Y.L., P.H.H. and W.H.C. designed the data analysis workflow, conducted genome assembly, and annotated the genome. P.C.H. and C.Y.L. prepared the initial manuscript draft, which was subsequently revised by C.Y.L., C.L.A.C. and S.H.C. All authors have reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hsu, PC., Lin, CY., Hsieh, PH. et al. Chromosome-Level Genome Assembly and Annotation of the Japanese Cutlassfish (Trichiurus japonicus): A High-Quality Genomic Resource Featuring Nuclear and Mitochondrial Completeness for Future Studies. Sci Data 12, 1860 (2025). https://doi.org/10.1038/s41597-025-06112-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06112-3
