
Abstract
Metagenomic Next-Generation Sequencing (mNGS) enables simultaneous sequencing of both microbial and host nucleic acids in clinical samples. However, analytical approaches for interpreting complex mNGS datasets are seldom disclosed, limiting advancements in multimodal analysis and omics-driven research models built upon mNGS results. We present 402 high-quality bronchoalveolar lavage fluid mNGS DNA and RNA sequencing datasets for developing combined microbial-host metagenomic diagnostic approaches. Only the microbial (non-host) sequence reads have been deposited. We provide comprehensive descriptions of methods, tools, and pipelines used for mining microbial features (DNA/RNA microbial composition and bacteriophage abundances) and host response features (differential expression genes, transposable elements, cell-type composition, and copy number variation). These data processing pipelines set a standard for future multimodal omics diagnostic research, promoting the adoption of standardized practices in omics-based studies that integrate clinical data.
Similar content being viewed by others

Background & Summary
Metagenomic next-generation sequencing (mNGS) is an emerging omics analysis technique capable of simultaneously sequencing microbial and host nucleic acid in clinical specimens. It has found increasingly widespread clinical applications in pathogen identification related to infectious diseases1,2. In addition to microbial sequences containing information about pathogens and microbiota (typically constituting less than 10% of sequencing reads), mNGS sequencing datasets also encompass vast amounts of host genomic sequences (often comprising over 90% of the total sequenced reads)3,4. These host sequences contain rich genomic features that can reflect the patient’s disease status, such as differentially expressed genes associated with immune-inflammatory responses5. Utilizing advanced bioinformatics tools to maximize the extraction of specific biological features relevant to the patient’s disease state from these complex sequencing data can aid in comprehensively understanding the pathophysiological mechanisms of the disease and developing reliable intelligent diagnosis and treatment strategies.
In recent years, a handful of studies have led the way in developing mNGS data-based omics diagnostic models. These models integrate pathogen metagenomics and host transcriptomics data from mNGS results of clinical samples like blood, cerebrospinal fluid, or respiratory specimens, targeting diagnoses for conditions such as sepsis, encephalitis, and acute respiratory diseases6,7,8. These efforts have yielded commendable performance in disease discrimination. However, there are currently no detailed design schemes or data analysis workflows disclosed for machine learning-based intelligent diagnostic strategies in microbial-host metagenomics. This limitation restricts the dissemination and replication of such research findings.
In 2023, our team successfully developed a microbial-host metagenomics-based intelligent differential diagnostic strategy for diagnosing lung cancer and pulmonary infections (bacterial, fungal, and tuberculosis infections) using bronchoalveolar lavage fluid (BALF) mNGS datasets9,10. Currently, we have optimized the diagnostic model and methodology, while simultaneously publicly releasing the mNGS datasets (unhost) of 402 patients used for model building (NCBI Sequence Read Archive under the BioProject ID: PRJNA105676511). To our knowledge, this is the largest high-quality BALF mNGS datasets currently available.
In this data descriptor, we provide a comprehensive overview of the methodologies and pipelines utilized in the extraction of microbial features (DNA/RNA microbial composition and bacteriophage abundances) and host response features (differential expression genes, transposable elements, cell-type composition, and copy number variation) from the 402 BALF mNGS datasets. These data processing pipelines will provide examples of data mining and integrated utilization for more similar multi-omics diagnostic studies in the future, thereby promoting the standardized practice of omics research based on clinical data.
Methods
The mNGS dataset in this study comprised 402 adult patients admitted to the First Affiliated Hospital, Zhejiang University School of Medicine (FAHZU) between 8 March 2020 and 27 May 2023. These patients were suspected of having lung cancer or pulmonary infections. Inclusion criteria required patients to be aged ≥18 years and to have undergone BALF sampling within 72 hours of intubation to identify causative pathogens. Exclusion criteria included underlying leukaemia, absence of a definitive diagnosis after extensive follow-up, or lack of matching DNA and RNA mNGS data from BALF samples. The diagnosis of lung cancer was based on clinical suspicion, supported by laboratory results from cytology, flow cytometry, and/or tissue biopsy. Pathological information for all samples was assessed according to the 2015 WHO Histological Classification of Lung Cancer and determined from surgically resected tissue sections. The diagnosis of pulmonary infections was based on clinical suspicion and confirmed through standard microbiological diagnostics, including cultures, antigen/antibody tests, PCR, and sequencing. This study retrospectively analyzed archival materials at FAHZU under a no-patient-contact research protocol, which was approved by the FAHZU Institutional Review Board (IIT20220714A). Prior to sample collection, written informed consent had been obtained from patients, covering the use of residual samples for research purposes. According to Guidance of the Ministry of Science and Technology (MOST) for the Review and Approval of Human Genetic Resources, we could not share sequencing data with homo sapiens.
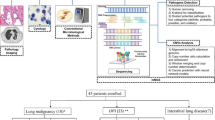
Here, we present a more condensed version of the methods fully described in Chen, Y. et al.12. The workflow is shown in Fig. 1. We make the raw sequencing data (unhost) freely available in NCBI Sequence Read Archive under the BioProject ID: PRJNA105676511, and scripts together with more downstream analysis results are accessible as the GitHub13.

Workflow of sample and data processing. Samples and data are shown in grey and processes highlighted in blue.
The procedure of collecting BALF samples
BALF was performed using flexible bronchoscopy under local anaesthesia. The bronchoscope was advanced to a radiologically involved lung segment, and 100–150 mL of sterile 0.9% saline was instilled in 20–50 mL aliquots. After each instillation, fluid was gently aspirated and pooled. BALF specimens were immediately stored on ice and processed within 2 hours. Samples with visible blood contamination or recovery <30% were excluded.
BALF DNA/RNA sequencing methods
Wet lab BALF sequencing methods were described in previous study12,14. In brief, we recruited 123 lung cancer cases, 279 cases of pulmonary infections including tuberculosis, fungal, and bacterial infections, and 32 negative control cases that include conditions like immune pneumonitis, organizing pneumonia, and drug-related pneumonia. For BALF DNA sequencing, we treated 1 mL of BALF samples with 1 U benzonase and 0.5% Tween 20, incubating at 37 °C for 5 minutes to deplete host nucleic acids. Subsequently, 600 µL of this mixture was subjected to bead beating with ceramic beads in a Minilys Personal TGrinder H24 Homogenizer, followed by nucleic acid extraction from 400 µL of the sample using a QIAamp UCP Pathogen Mini Kit, with the final DNA elution in 60 µL. DNA quantity was assessed using a Qubit dsDNA HS Assay Kit. For BALF RNA sequencing, 1 mL BALF samples were centrifuged, and the precipitate was processed with TRIzol LS for RNA extraction using a Direct-zol RNA Miniprep kit. Library preparations for sequencing involved using 30 µL of DNA with the Nextera DNA Flex kit and 10 µL of purified RNA with the Ovation Trio RNA-Seq Library Preparation Kit. Library concentrations were quantified using a Qubit dsDNA HS Assay Kit, quality assessed via an Agilent 2100 Bioanalyzer with a High Sensitivity DNA kit, and sequencing performed on an Illumina NextSeq. 550 sequencer employing a 50-cycle single-end strategy2,3,4,15.
Generating microbial and host expression matrix
Detailed pipeline information for microbial and gene expression profiling is available at GitHub13. All parameters and database we used were showed with shell scripts. In short, we utilized a validated mNGS protocol aimed at comprehensive microbial composition analysis15,16,17. The process initiated with the use of fastp18 for the elimination of low-quality reads, duplicates, sequences shorter than 50 base pairs, and adapter contamination. To remove human genetic sequences, reads were aligned against the hg38 human reference genome using BWA (0.7.17)19. The generation of taxonomic profiles was facilitated by Kraken2 v2.0.7 and Bracken v2.5, which operated under default settings and employed a widely recognized database. To account for differences in sequencing depth, the sequencing reads identifying microbes were normalized to reads per million (RPM)20. Host gene expression was analyzed by aligning high-quality data to the human genome via HISAT221, using default settings, with gene-level quantification conducted through featureCounts. The aggregate gene counts were compiled using the featureCounts utility from the Subread package release 2.0.022.
In our GitHub repository and figshare (https://doi.org/10.6084/m9.figshare.29388539.v1)13, the kraken2_pipeline folder contains the following scripts for generating the microbial abundance matrix:
-
01_data_preprocessing.sh: uses fastp to remove low-quality reads and adapter sequences.
-
02_rmhost.sh: uses BWA and samtools to remove host reads by aligning to the human reference genome (hg38).
-
03_kraken2_bracken.sh: uses Kraken2 and Bracken to perform microbial taxonomic profiling.
-
04_relative_abudances_matrix.sh: calculates the relative abundance of each microbial taxon across all samples and summarizes the results at both the species and genus levels.
The RNAseq_pipeline folder includes scripts for read mapping, gene expression quantification, and profile generation:
-
01_data_preprocessing.sh: performs the same preprocessing as in the kraken2_pipeline using fastp.
-
02_map_to_reference_hg38.sh: uses HISAT2 to align reads to the hg38 reference genome.
-
03_featureCount.sh: uses featureCounts to generate gene-level read count matrices for individual samples. A specific version of the gene annotation file (GFF format) is provided to ensure result reproducibility.
-
04_readcount_to_expression_profiles.sh: merges the read count tables from all samples into a single gene expression matrix.
Microbial de-contamination
As previously reported in our methodology15,23, all wet-lab experiments were conducted under strict sterile conditions. Negative controls (PBS or sterile water) were included during nucleic acid extraction and library preparation to monitor potential contamination. These controls were processed in parallel with clinical samples throughout the experimental workflow. Our bioinformatic pipeline incorporated multiple layers of contamination control: ①Host DNA removal: Sequences were aligned to the human reference genome (GRCh38) and filtered. ②Decontam package application: We utilized the prevalence-based mode of the Decontam R package (v1.12.0), which statistically identifies contaminant taxa by comparing sequence frequencies between samples and negative controls. After generating microbial matrix, we used negative control to filter microorganisms which might be highly potentials as contamination. However, this step was quite independent for different laboratories. Thus, we provided our method as an optional procedure. First, negative controls were derived from BALF mNGS datasets of several (in our studies, there were 32, microbiological results and diagnosis of 32 negative control patients were listed in Table S3) individuals without infection or cancer, against which microbial abundance was compared. We then determined the mean and standard deviation of species’ relative abundances within these controls, establishing a threshold for positive detection at the mean plus three standard deviations. Second, microbes exceeding this threshold in the mNGS datasets of patients with lung cancer or infections were identified as ‘positive’ and included in our following microbial count analysis15.
Other omics information
We conducted secondary bioinformatic analysis employing various software tools. Firstly, we estimated the abundances of Transposable Elements (TE) using TEtranscripts24 and performed differential expression analysis with default parameters. Secondly, we identified immune-related genes (IRGs) using data from the ImmPort database (https://www.immport.org/home), and interferon-stimulated genes (ISGs) sourced from a referenced study25. Thirdly, to estimate the relative proportions of immune cells, we quantified transcript levels in TPM and employed digital cytometry via CIBERSORTx with the original gene signature file LM22 and 1000 permutations26. Fourthly, we identified tumor fractions or copy number variants through ichorCNA27, CNVkit28, and estimate29, adhering to the software instructions. Lastly, for bacteriophage annotation, we aligned cleaned reads against a curated phage database (CPD) using blastn30. Detailed parameters for each software or pipeline are outlined in our preprint manuscript12.
Differentially expressed genes (DEGs) and TE were identified in each group using the DESeq. 2 package, applying criteria of FDR ≤0.05 and Fold-change ≥1.531. Gene set enrichment analysis (GSEA) for DEGs was carried out using the REACTOME, KEGG, and GO databases by the fgsea package32,33,34. Significantly enriched pathways or biological processes were determined based on Fisher’s exact test (p-value < 0.05), following Benjamini-Hochberg adjustment. Latent variables were calculated by PLIER R package35. Wilcoxon rank-sum test assessed the difference between each group’s probability value.
Data Records
Microbial reads (unhost) from metagenomic and metatranscriptomic data were deposited in NCBI Sequence Read Archive under the BioProject ID: PRJNA105676511. Detailed metadata for all samples (402 cohort samples, 32 DNA negative controls, and 32 RNA negative controls) are provided in Supplementary Table S1 (DNA metadata) and Supplementary Table S2 (RNA metadata). These tables include information such as group classification, BioSample accession number, SRA accession number, total reads, total bases, number of clean reads, clean read rate (%), and other relevant details. According to Guidance of the Ministry of Science and Technology (MOST) for the Review and Approval of Human Genetic Resources, we could not share sequencing data with homo sapiens. Thus, host gene expression profile derived from metatranscriptomic were deposited in NCBI GEO datasets36. For other scientific studies and reproducibility of our results, data resulting from the matrix generation presented here are available on Figshare (https://doi.org/10.6084/m9.figshare.29388539.v1), which contains the processed microbial abundance matrices and expression tables. The corresponding analysis scripts and pipelines remain available in the GitHub repository13. The data are separated into several subfolders:
-
The folder contains raw read counts in subfolder /data/RNA_EXP, count-based expression profiles of all samples stored in file RNAseq_featurecount.zip. These files are the main outcomes of gene expression profile classifications.
-
The folder contains raw read counts in subfolder /data/TE_EXP, count-based expression profiles of all samples stored in fileTransposable_element_count_table.zip. These files are the main outcomes of Transposable element expression profile called by TEtranscripts.
-
Results of copy number variants and tumour fraction derived from it are located in the folder /data/CNV with one table contain all results called by ichorCNV,CNVkit and estimate.
-
Results of the metagenomics analysis of the non-human genomic content of DNA-seq RNA-seq are located in folder /data/DNA_kraken2 and /data/RNA_kraken2 with two tables (*__abundances.txt) containing lists of bacterial species used in this metagenomics analysis. Also, species and genus relative abundance table of negative controls were also uploaded separately on each folder.
Data Overview
We analyzed the microbial composition at both the genus and species levels based on DNA sequencing data from patients with different pulmonary diseases (Fig. 2A and B). At the genus level, the three most abundant genera were Pseudomonas, Streptococcus, and Prevotella, which are common commensals in the human respiratory tract. Notably, Pseudomonas was highly prevalent in samples from the bacterial pneumonia group, Aspergillus in the fungal pneumonia group, and Mycobacterium in the tuberculosis group—organisms known to be pathogens associated with these disease categories (Fig. 2A). At the species level, we observed similar results. Pseudomonas aeruginosa, Rothia mucilaginosa, Neisseria mucosa, and Klebsiella pneumoniae showed high relative abundances in many samples. In particular, Pseudomonas aeruginosa (bacterial group), Mycobacterium tuberculosis (TB group), and Aspergillus fumigatus (fungal group) were significantly enriched in their respective pneumonia patient subgroups (Fig. 2B).

Microbial composition of DNA sequencing data in species and genus levels. (A) Top genus of microbial composition among different pulmonary diseases (Bacteria, Bacterial pneumonia; Fungi, Fungal pneumonia; TB, Pulmonary Tuberculosis; Cancer, Lung Cancer). (B) Top species of microbial composition among different pulmonary diseases (Bacteria, Bacterial pneumonia; Fungi, Fungal pneumonia; TB, Pulmonary Tuberculosis; Cancer, Lung Cancer).
Technical Validation
The quality of the sequencing data was evaluated through multiple layers of quality control and validation to ensure the integrity and reproducibility of the dataset. The raw reads from both DNA-seq and RNA-seq experiments were first assessed using FASTQC, and representative per-base quality plots are shown in Fig. 3A (DNA-seq) and Fig. 3B (RNA-seq). Regardless of the initial data quality, all samples underwent standardized data cleaning to ensure that no base was called with a Phred quality score below 20.

Per base quality of raw sequencing data. Output of multiQC: (A) DNA sequencing of 402 BAL samples; (B) RNA sequencing of 402 BALF samples.
Summary statistics of sequencing performance are provided in Table 1 and Table 2, showing comparable data yields and quality metrics across the four clinical subgroups (bacterial, fungal, tuberculosis, and lung cancer cohorts). The average clean read counts ranged between approximately 19–24 million reads per sample, and clean read rates exceeded 98% for all groups, indicating stable sequencing performance.
To verify the clinical accuracy and consistency of the dataset, each case was reviewed and confirmed through standard diagnostic criteria. Diagnoses of pulmonary infections (bacterial, fungal, and tuberculosis) were based on clinical evaluation combined with conventional microbiological methods (culture, PCR, and antigen/antibody testing). Lung cancer diagnoses were established by cytology, flow cytometry, and/or tissue biopsy, following the 2015 WHO Histological Classification of Lung Tumors.
Together, these quality control procedures demonstrate that the sequencing data and clinical annotations are of high integrity and suitable for reuse in future methodological and computational studies.
Data availability
The metagenomic sequencing datasets generated and analyzed in this study have been deposited in the NCBI database. DNA-seq and RNA-seq data are available under BioProject accession number PRJNA1056765 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1056765). The corresponding processed transcriptomic data are available in the Gene Expression Omnibus (GEO) under accession number GSE252118 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE252118).
Code availability
Our study utilizes an AMD EPYC 7763 dual-socket server configuration featuring 128 physical cores and 1TB of RAM, complemented by 50TB of storage. All the code used to process the genomic data is freely available as a part of the provided GitHub repository13 and the code is located in the folder named scripts. The scripts folder also contains dependencies and the used parameter values. Depending on the scripts’ functionality, they are separated into various folders:
• The folder RNAseq_pipeline contains scripts used for read mapping, gene expression quantification, and profile classification.
• The folder kraken2_pipeline contains scripts to assign reads to bacterial taxa and the scripts for differential analysis of bacterial species among pulmonary diseases.
• The folder classifier modelling contains scripts and input files for machine learning of pulmonary disease classifier building.
References
Han, D. et al. The Real-World Clinical Impact of Plasma mNGS Testing: an Observational Study. Microbiol Spectr 11(2), e0398322 (2023).
Han, D. et al. Applicability of Bronchoalveolar Lavage Fluid and Plasma Metagenomic Next-Generation Sequencing Assays in the Diagnosis of Pneumonia. Open Forum Infect Dis 11(1), ofad631 (2024).
Han, D. et al. Multilaboratory assessment of metagenomic next-generation sequencing for unbiased microbe detection. J Adv Res 38, 213–22 (2022).
Diao, Z., Han, D., Zhang, R. & Li, J. Metagenomics next-generation sequencing tests take the stage in the diagnosis of lower respiratory tract infections. J Adv Res 38, 201–12 (2022).
Chiu, C. Y. & Miller, S. A. Clinical metagenomics. Nat Rev Genet 20(6), 341–55 (2019).
Ramachandran, P. S. et al. Integrating central nervous system metagenomics and host response for diagnosis of tuberculosis meningitis and its mimics. Nat Commun 13(1), 1675 (2022).
Langelier, C. et al. Integrating host response and unbiased microbe detection for lower respiratory tract infection diagnosis in critically ill adults. Proc Natl Acad Sci USA 115(52), E12353–e62 (2018).
Kalantar, K. L. et al. Integrated host-microbe plasma metagenomics for sepsis diagnosis in a prospective cohort of critically ill adults. Nat Microbiol 7(11), 1805–16 (2022).
Han D, Yu F, Chen Y. A-242 Integrating Respiratory Metagenomics and Metatranscriptomics for Diagnosis of Lung Cancer and Infection in Patients with Pulmonary Diseases. Clinical Chemistry 2023; 69(Supplement_1).
Han D. Integrating Respiratory Metagenomics and Metatranscriptomics for Diagnosis of Lung Cancer and Infection in Patients with Pulmonary Diseases. American Association of Clinical Chemistry (AACC) Annual Scientific Meeting (https://www.myadlmorg/community/academy-of-diagnostics-and-laboratory-medicine/awards-and-events/distinguished-abstracts-winners-2023) (2023).
National Center for Biotechnology Information. “Sequence Read Archive accession SRP480031.” NCBI, 2024, identifiers.org/ncbi/insdc.sra:SRP480031. Accessed 26 June 2025.
Chen Y, et al. Metagenomic Analysis of Bronchoalveolar Lavage Fluid Enables Differential Diagnosis Between Lung Cancer and Pulmonary Infections 2024.
Yang B. Bronchoalveolar lavage fluid mNGS DNA and RNA sequencing datasets and analysis script. figshare. Dataset. https://doi.org/10.6084/m9.figshare.29388539.v1 (2025).
Zhang, X. et al. Diagnosis of Non-Tuberculous Mycobacterial Pulmonary Disease by Metagenomic Next-Generation Sequencing on Bronchoalveolar Lavage Fluid. Infect Drug Resist 16, 4137–45 (2023).
Diao, Z. et al. Validation of a Metagenomic Next-Generation Sequencing Assay for Lower Respiratory Pathogen Detection. Microbiol Spectr 11(1), e0381222 (2023).
Sulaiman, I. et al. Microbial signatures in the lower airways of mechanically ventilated COVID-19 patients associated with poor clinical outcome. Nat Microbiol 6(10), 1245–58 (2021).
Zhou, Z. et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe 27(6), 883–90.e2 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17), i884–i90 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14), 1754–60 (2009).
Lu, J. et al. Metagenome analysis using the Kraken software suite. Nat Protoc 17(12), 2815–39 (2022).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37(8), 907–15 (2019).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30(7), 923–30 (2014).
Tao, Y. et al. Diagnostic Performance of Metagenomic Next-Generation Sequencing in Pediatric Patients: A Retrospective Study in a Large Children’s Medical Center. Clin Chem 68(8), 1031–41 (2022).
Jin, Y., Tam, O. H., Paniagua, E. & Hammell, M. TEtranscripts: a package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 31(22), 3593–9 (2015).
Schoggins, J. W. et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472(7344), 481–5 (2011).
Steen, C. B., Liu, C. L., Alizadeh, A. A. & Newman, A. M. Profiling Cell Type Abundance and Expression in Bulk Tissues with CIBERSORTx. Methods Mol Biol 2117, 135–57 (2020).
Adalsteinsson, V. A. et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun 8(1), 1324 (2017).
Talevich, E., Shain, A. H., Botton, T. & Bastian, B. C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol 12(4), e1004873 (2016).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4, 2612 (2013).
Haddock, N. L. et al. Phage diversity in cell-free DNA identifies bacterial pathogens in human sepsis cases. Nat Microbiol 8(8), 1495–507 (2023).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biol 15(12), 550 (2014).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102(43), 15545–50 (2005).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45(D1), D353–d61 (2017).
Gillespie, M. et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res 50(D1), D687–d92 (2022).
Mao, W., Zaslavsky, E., Hartmann, B. M., Sealfon, S. C. & Chikina, M. Pathway-level information extractor (PLIER) for gene expression data. Nat Methods 16(7), 607–10 (2019).
Han D. GEO https://identifiers.org/geo/GSE252118.
Acknowledgements
We thank all clinicians who provided detailed testing, diagnostic and treatment data of patients for our study, as well as all infectious disease (ID) physicians, clinical microbiologists and oncologists who received our clinical consultations. Three funding sources: “Leading Geese” Research and Development Plan of Zhejiang Province (No. 2024C03218), National Natural Science Foundation of China (No. 82472371), National Key R&D Program of China (2023YFC2308300).
Author information
Authors and Affiliations
Contributions
H.T. and H.L. collected clinical data and contributed to manuscript writing and editing. L.Y., M.X., J.Z., J.W., and J.Z. performed nucleic acid extraction and sequencing preparation of BALF samples. H.L., B.Y., and D.H. revised the manuscript based on the reviewers’ comments. B.L. and D.H. designed the study, participated in clinical data analysis, and drafted and edited the primary manuscript. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tang, H., Liu, H., Yuan, L. et al. Bronchoalveolar lavage fluid metagenomic datasets: a multidimensional clinical biomolecular resource. Sci Data 12, 1919 (2025). https://doi.org/10.1038/s41597-025-06171-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06171-6
