
Abstract
The white-spotted sawyer, Monochamus scutellatus (Say) (Coleoptera: Cerambycidae), is an important vector of pinewood nematode (PWN), Bursaphelenchus xylophilus (Steiner and Buhrer) Nickle, in North America. While Monochamus species from the Palearctic region have been extensively studied for their role in transmitting PWN that causes pine wilt disease in Asia and Europe, the genetic mechanisms underlying Monochamus-PWN interactions in their native range remain largely unknown. Here, we present the first chromosome-level genome assembly of the North American M. scutellatus, constructed using PacBio HiFi long read, Pore-C chromatin conformation capture, and Illumina RNA sequencing. The assembled genome spans 830.9 Mbp, with a scaffold N50 of 87.9 Mbp, 97.9% of which were anchored to 10 chromosome-level scaffolds. The X chromosome was identified through synteny analysis. Repeat elements constitute 70.7% of the genome, and 13,684 protein-coding genes were functionally annotated. This reference-quality genome of M. scutellatus provides a valuable comparative resource for elucidating the genomic basis of Monochamus-PWN interactions, and offers a foundation for devising targeted management strategies against PWN and its vectors.
Similar content being viewed by others

Background & Summary
Longhorned beetles (family Cerambycidae) represent one of the most species-rich families of beetles, with over 36,000 described species in 4,100 genera worldwide1,2. As a major lineage of phytophagous beetles, most longhorned beetles attack and feed on plant tissues, and their diversification is often explained by a coevolutionary radiation with diversifying angiosperms3,4,5. Among them, the Asian longhorned beetle, Anoplophora glabripennis (Motschulsky), was one of the first species to be investigated for the genetic basis of plant-feeding evolution, due to its broad host range and invasive pest status in the United States6,7,8. Recent transcriptomic and genomic studies of longhorned beetles, including a reference genome of A. glabripennis, have revealed the presence of horizontally acquired plant cell wall-degrading enzymes in the glycoside hydrolase families, which likely facilitate nutrient acquisition from nutrient-poor, recalcitrant woody tissues6,9,10,11. More recently, chromosome-level genome assemblies have also been generated for two other important xylophagous longhorned beetles, Monochamus alternatus (Hope) and M. saltuarius (Gebler, 1830)12,13—major vectors of pinewood nematodes in East Asia—providing an unparalleled opportunity to study the genomic basis of conifer-feeding and adaptive traits associated with life in temperate forests.
Monochamus longhorned beetles are distributed worldwide, except Australasia, and include a monophyletic clade of 18 conifer-feeding species restricted to temperate forests of the Holarctic region. These conifer specialists are inferred to have evolved within a predominantly angiosperm-feeding lineage of Monochamus at the Miocene-Pliocene boundary around 5 million years ago (Mya)14. In fact, most of these conifer-feeding species are known to transmit the pinewood nematode (PWN), Bursaphelenchus xylophilus (Steiner and Buhrer) Nickle (Nematoda: Aphelenchoididae), the causal agent of pine-wilt disease in the Palearctic region15,16,17 and a nematode species native to North America. While the biology and pest control measures for Palearctic Monochamus species—such as Monochamus alternatus and M. saltuarius—have been extensively studied, the genetic mechanisms underlying Monochamus-PWN interactions in their native North American range remains largely unexplored.
In this study, we present the first chromosome-level genome assembly of Monochamus scutellatus (Say), a major vector of PWN in North America18, generated based on PacBio HiFi long reads, Pore-C chromatin confirmation capture, and Illumina RNA-seq data. The genome spans 830.9 Mbp and comprises 10 pseudo-chromosomes (Fig. 2A,B; Table 2), consistent with previous cytological evidence19. Chromosome 10 was identified as the X chromosome based on synteny analysis, which revealed extensive conservation of the X chromosome across Coleoptera (Fig. 2C). With a genome size comparable to those of the two Palearctic congeners—M. alternatus (792.1 Mbp) and M. saltuarius (682.2 Mbp), the M. scutellatus genome demonstrates exceptional contiguity, reflected by fewer scaffolds and a higher N50 (Table 2). As the first genomic resource for a North American Monochamus species, the M. scutellatus genome provides a valuable foundation for investigating the genomic basis of Monochamus-PWN interactions in their region of origin and offers a key comparative framework for testing evolutionary hypotheses on the origin of these interactions, as well as their role in the beetles’ adaptation to utilizing the vast resource of coniferous forests across the Northern Hemisphere.
Methods
Sample collection
Adult specimens of Monochamus scutellatus were collected in July 2023 from Eastern white pine, Pinus strobus Linnaeus (Pinaceae), at Blue Hills Reservation, Milton, Massachusetts, U.S.A. (42°13.237′N, 71°7.037′W; elev. 81 m), using panel traps equipped with monochamol pheromone lures (Fig. 1). To minimize contamination from gut contents, all specimens were starved for several days, flash-frozen in liquid nitrogen, and subsequently cryo-preserved at −80 °C until used for extraction. A total of two female specimens were used: One for PacBio sequencing and the other for Pore-C and Illumina transcriptome sequencing. The voucher specimen for PacBio sequencing (voucher no.: MCZ-SK1313) has been deposited in the Entomology Research Collection at the Museum of Comparative Zoology, Harvard University.

(A) Voucher specimen of Monochamus scutellatus (MCZ-SK1313; female) used for genome sequencing. (B) Habitat of M. scutellatus consisting of Eastern white pine (Pinus strobus), from which specimens were collected using panel traps equipped with monochamol pheromone lures (Milton, Massachusetts, USA).
Nucleic acid extraction and sequencing
High molecular weight (HMW) genomic DNA (gDNA) was extracted from the thoracic muscle tissue of an individual adult specimen using the Qiagen MagAttract HMW DNA Kit (Qiagen, Hilden, Germany). The integrity of the extracted gDNA was evaluated via gel electrophoresis on a 1% agarose gel with a lambda DNA marker, while concentration and purity were assessed using a Quantus Fluorometer (Promega, Madison, WI, USA) and a Nanodrop Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Purified HMW gDNA was treated with the Short Read Eliminator (SRE) XL Kit (Pacific Biosciences, Menlo Park, CA, USA) to remove DNA fragments below 40 kbp, and sheared into 20 kbp fragments using the Megaruptor 2 (Diagenode, Liège, Belgium). A PacBio SMRT library was constructed using the SMRTbell Prep Kit 3.0, and sequenced on a single SMRT HiFi cell of the PacBio Sequel IIe system at the National Instrumentation Center for Environmental Management (NICEM), Seoul National University (Seoul, Republic of Korea), generating 34.0 Gbp of HiFi reads (Table 1).
Chromatin conformation capture sequencing was performed on half of a longitudinally bisected specimen (voucher no.: MCZ-SK1314) following the Pore-C protocol20. Briefly, chromatin was fixed in situ within intact nuclei using formaldehyde to preserve native 3-D interactions. Following permeabilization of the nuclei, chromatin was denatured to expose accessible regions and digested with the restriction enzyme NlaIII (New England Biolabs, Ipswich, MA, USA). Proximally crosslinked DNA fragments were then ligated, and purified via phenol:chloroform extraction. The final Pore-C library was prepared using the Genomic DNA by Ligation Protocol [SQK-LSK114; Oxford Nanopore Technologies (ONT), Oxford, UK], and sequenced on a single flowcell of the PromethION system at NICEM (Seoul, Republic of Korea), yielding 22.5 Gbp of Pore-C reads with Phred Q-score ≥10 (Table 1).
Total RNA was extracted from the remaining half of the second specimen using the mirVana miRNA Isolation Kit (Invitrogen, Waltham, MA, USA). RNA concentration and integrity were evaluated using a Nanodrop Spectrophotometer and an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). An mRNA library was constructed using the NEBNext Ultra II RNA Library Prep Kit (New England Biolabs, Ipswich, MA, USA) and sequenced on a 150-bp paired-end S4 flowcell of the NovaSeq 6000 platform (Illumina, San Diego, CA, USA) at Novogene (Sacramento, CA, USA), producing 17.2 Gbp of Illumina RNA-seq reads (Table 1).
Genome assembly and scaffolding
To assemble the genome of M. scutellatus, genome size and heterozygosity were first estimated from the raw PacBio reads using Jellyfish v2.3.021 and GenomeScope v2.022 with a k-mer size of 35, which estimated the genome size of 774.11 Mbp and heterozygosity of 1.21%. Primary contigs were assembled using Hifiasm v0.16.123, and haplotypic duplications were resolved by reassigning allelic contigs using Purge Haplotigs v1.1.224. The primary contigs were screened for potential contamination using BlobTools v1.1.125, based on which two of the 33 primary contigs that were identified as prokaryotic or having atypical GC content for arthropods were removed.
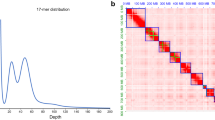
Chromosome-level scaffolds were constructed from the primary contigs and Pore-C data using the Pore-C Snakemake workflow20 and the 3D-DNA pipeline v18099226. The Hi-C contact map was visualized and manually curated in Juicebox v2.20.0027, and scaffolds were finalized with 3D-DNA. Scaffolding was further refined with RagTag v2.1.028 using the primary contigs as reference, increasing the mean scaffold lengths from 16.6 Mbp to 27.7 Mbp. Error correction was performed with Inspector v1.3.129 using the original raw PacBio HiFi reads. The final genome assembly was 830.9 Mbp in total length, slightly larger than the estimated genome size, 97.9% of which were assembled into 10 chromosome-scale scaffolds ranging from 152.6 Mbp to 28.4 Mbp (Table 2). A high-resolution Hi-C contact frequency heatmap was generated using HiGlass v0.8.030 to visualize chromosomal architecture (Fig. 2A). The mitochondrial genome was assembled using MitoHiFi v3.231, guided by the mitochondrial genome of Anoplophora glabripennis (GenBank accession: NC_008221.1) as a reference, and the final mitochondrial contig was selected based on annotations from MitoFinder v1.4.132.

Genomic characteristics of the Monochamus scutellatus genome. (A) Hi-C contact frequency heatmap (Pore-C) of the M. scutellatus genome, showing chromosomal interactions and scaffold continuity across the 10 pseudo-chromosomes. (B) Circular genome plot of the M. scutellatus genome, with inner tracks depicting key genomic features. (C) Time-calibrated phylogeny and macrosynteny across the genomes of M. scutellatus and other beetles, including six Cerambycidae and Tribolium castaneum (Tenebrionidae) as the outgroup. Chromosome nomenclature for reference species corresponds to their respective NCBI genome assemblies.
Repeat elements and gene annotations
Repeat regions and transposable elements (TE) in the M. scutellatus genome were predicted and annotated using both homology-based and de novo prediction approaches within the Earl Grey pipeline v4.4.033. RepeatMasker v4.1.534 was used to annotate repeats based on the Dfam v3.835 TE database for Coleoptera, and RepeatModeler v2.0.536 was employed to generate a species-specific de novo repeat library for M. scutellatus. De novo consensus TE sequences were curated through the “BLAST, Extract, Extend and Trim” (BEAT) process37, and long terminal repeat (LTR) retrotransposons were further annotated using LTR_Finder v1.0738. Repetitive elements accounted for 70.7% of the genome, with unknown repeats and DNA transposons comprising 41.1% and 30.4% of all repeats, respectively (Table 3).
Gene prediction was performed on the repeat-masked genome assembly using BRAKER v3.0.739, integrating species-specific transcriptomic and protein data, the Arthropoda OrthoDB, and reference protein datasets from Anoplophora glabripennis (GCF_000390285.2), Tribolium castaneum (GCF_000002335.3), Drosophila melanogaster (GCF_000001215.4) and Bombyx mori (GCF_030269925.1). A species-specific protein dataset for M. scutellatus was generated by assembling Illumina RNA-seq reads with rnaSPAdes v3.13.040, and translating RNA contigs into amino acid sequences with TransDecoder v5.7.041. Prior to the assembly, raw Illumina RNA-seq reads were adapter-trimmed and quality-filtered to a minimum Phred Q-score of 33 using Trimmomatic v0.3942. Ab initio gene prediction was conducted using AUGUSTUS v3.5.543, trained with transcriptome-based evidence from GenMark-ET v4.7244 and protein-based evidence from GeneMark-EP + v4.7245. Consensus gene models were generated by merging the outputs of seven gene prediction runs using TSEBRA v1.1.146, retaining only the longest isoform per gene. The final gene annotation was formatted into GFF using gFACs v1.1.247, resulting in a total of 21,110 predicted protein-coding genes (Table 2). Functional annotation of the predicted gene models was subsequently performed using eggNOG-mapper v2.1.1248 based on the Insecta eggNOG database, yielding a total of 13,684 genes functionally annotated (Tables 2, 4), with a BUSCO (Benchmarking Universal Single-copy Orthologs) protein completeness score of 97.6% (Table 5).
Synteny-based identification of the X chromosome
Genome synteny was analyzed across chromosome-level genome assemblies of seven Cerambycidae species, with Tribolium castaneum (Tenebrionidae) as an outgroup. Orthologous genes were identified via reciprocal-best BLASTp49 hits among annotated proteins using DIAMOND v2.0.1350. Chromosomal homology was evaluated through Bonferroni-corrected one-sided Fisher’s exact tests implemented in odp v0.3.351, based on the reciprocal-best BLASTp results. Conserved macrosyntenic blocks were visualized as ribbon diagrams. The analysis revealed extensive conservation of the X chromosome across all seven Cerambycidae species and T. castaneum, consistent with previous reports of high X chromosome conservation in Coleoptera12, and permitted the identification of chromosome 10 in the M. scutellatus genome as the X chromosome.
Orthologous gene identification and phylogenomic inference
Orthologous genes and orthogroups across the seven Cerambycidae and T. castaneum genomes were identified using OrthoFinder v2.5.552, with DIAMOND for sequence alignment and FastTree v2.1.1153 for maximum likelihood (ML) tree inference. A total of 3,708 single-copy orthologs were identified, aligned with MAFFT v7.5.2654, and trimmed using trimAl v1.455 with the gappyout algorithm. ML gene trees were inferred using IQ-TREE v2.2.2.656 with the MFP + MERGE option. A species tree was reconstructed under the multispecies coalescent model in ASTRAL v5.7.857, with T. castaneum as the outgroup. Divergence times were estimated within a Bayesian framework in MCMCtree, implemented in PAML v4.10.758, employing the approximate likelihood calculation method and two calibration points: (1) a fossil calibration for the crown-group Cerambycidae, based on the age of †Cretoprionus liutiaogouensis Wang et al. from the lower Cretaceous circa 122.5–124.0 Mya59; and (2) a secondary calibration for the Cerambycidae-Tenebrionidae divergence at approximately 220.2 Mya [95% highest posterior density (HPD): 188.1–237.6 Mya]5. The resulting time-calibrated phylogeny supports a sister-group relationship between M. scutellatus and the Palearctic clade comprising M. alternatus and M. saltuarius, which diverged approximately 11.8 Mya (95% HPD: 8.0–16.3 Mya) (Fig. 2C), providing robust evidence for the systematic placement and divergence history of M. scutellatus within Cerambycidae.
Data Records
All raw sequencing data (PacBio HiFi, ONT Pore-C, and Illumina RNA-seq) used for genome assembly and annotation for M. scutellatus have been deposited in NCBI BioProject PRJNA1289024. PacBio HiFi sequencing data, ONT Pore-C sequencing data and Illumina RNA-seq data are available within the NCBI Sequence Read Archive (SRA) under accession numbers SRR3444437960, SRR3444437861, and SRR3444437762, respectively. The final chromosome-level genome assembly has been deposited in GenBank under accession number GCA_052862855.163. The genome assembly and annotation files are available from the Figshare Repository (https://doi.org/10.6084/m9.figshare.2957536164).
Technical Validation
The final genome assembly, constructed using PacBio HiFi and Pore-C sequencing data, along with transcriptome data, was assembled to 10 chromosome-level scaffolds. Genome completeness was assessed with BUSCO v5.8.065 against the Insecta OrthoDB v1066 and revealed 99.0% of core single-copy orthologs, with only 0.8% of duplicated genes (Table 5), exceeding the 90% BUSCO threshold recommended for reference genomes67. To further evaluate assembly quality, raw PacBio HiFi reads were mapped back to the final genome assembly using Minimap v2.2168 within Inspector, resulting in a mapping rate of 99.6%, an average alignment depth of 40.7×, and an assembly quality value (QV) of 36.9 (Table 6), indicating a highly complete and contiguous assembly.
Data availability
All datasets are available through the NCBI SRA (https://www.ncbi.nlm.nih.gov/sra) under accession numbers SRR34444377, SRR34444378 and SRR34444379; the NCBI GenBank (https://identifiers.org/ncbi/insdc.gca:GCA_052862855.1); and the Figshare Repository (https://doi.org/10.6084/m9.figshare.29575361).
Code availability
All software and pipelines were executed according to the manuals provided by the published bioinformatics tools. The version of each program is provided in the Methods section, and default parameters were used unless otherwise stated. No custom scripts were used.
References
Svacha, P. & Lawrence, J. F. Cerambycidae Latreille, 1802. in Handbook of Zoology, Arthropoda: Insecta; Coleoptera, Beetles, Volume 3: Morphology and systematics (Phytophaga) (eds Richard A. B. Leschen & Rolf G. Beutel) 77–177 (Walter de Gruyter, 2014).
Monné, M. L., Monné, M. A. & Wang, Q. General morphology, classification, and biology of Cerambycidae. in Cerambycidae of the world: Bioogy and pest management (ed Qiao Wang) 1–70 (CRC Press, 2017).
Farrell, B. D. “Inordinate fondness” explained: Why are there so many beetles? Science 281, 555–559 (1998).
Farrell, B. D. & Mitter, C. The timing of insect/plant diversification: Might Tetraopes (Coleoptera: Cerambycidae) and Asclepias (Asclepiadaceae) have co-evolved? Biological Journal of the Linnean Society 63, 553–577 (1998).
McKenna, D. D. et al. The evolution and genomic basis of beetle diversity. Proceedings of the National Academy of Sciences of the United States of America 116, 24729–24737 (2019).
McKenna, D. D. et al. Genome of the Asian longhorned beetle (Anoplophora glabripennis), a globally significant invasive species, reveals key functional and evolutionary innovations at the beetle–plant interface. Genome Biology 17, 227–227 (2016).
Scully, E. D. et al. Functional genomics and microbiome profiling of the Asian longhorned beetle (Anoplophora glabripennis) reveal insights into the digestive physiology and nutritional ecology of wood feeding beetles. BMC genomics 15, 1096–1096 (2014).
Scully, E. D. et al. Metagenomic profiling reveals lignocellulose degrading system in a microbial community associated with a wood-feeding beetle. PLoS ONE 8, 1–22 (2013).
Kirsch, R. et al. Horizontal gene transfer and functional diversification of plant cell wall degrading polygalacturonases: Key events in the evolution of herbivory in beetles. Insect Biochemistry and Molecular Biology 52, 33–50 (2014).
Shin, N. R. et al. Larvae of longhorned beetles (Coleoptera; Cerambycidae) have evolved a diverse and phylogenetically conserved array of plant cell wall degrading enzymes. Systematic Entomology 46, 784–797 (2021).
Shin, N. R., Doucet, D. & Pauchet, Y. Duplication of horizontally acquired GH5-2 enzymes played a central role in the evolution of longhorned Beetles. Molecular Biology and Evolution 39, 1–14 (2022).
Fu, N. et al. Chromosome-level genome assembly of Monochamus saltuarius reveals its adaptation and interaction mechanism with pine wood nematode. International Journal of Biological Macromolecules 222, 325–336 (2022).
Gao, Y. F. et al. Chromosome-level genome assembly of the Japanese sawyer beetle Monochamus alternatus. Scientific Data 11, 199 (2024).
Gorring, P. S. & Farrell, B. D. Evaluating species boundaries using coalescent delimitation in pine-killing Monochamus (Coleoptera: Cerambycidae) sawyer beetles. Molecular Phylogenetics and Evolution 184, 107777 (2023).
Tóth, Á. Bursaphelenchus xylophilus, the pinewood nematode: Its significance and a historical review. Acta Biologica Szegediensis 55, 213–217 (2011).
Akbulut, S. & Stamps, W. T. Insect vectors of the pinewood nematode: A review of the biology and ecology of Monochamus species. Forest Pathology 42, 89–99 (2012).
Vicente, C., Espada, M., Vieira, P. & Mota, M. Pine wilt disease: A threat to European forestry. European Journal of Plant Pathology 133, 89–99 (2012).
Wingfield, M. J. & Blanchette, R. A. The pine-wood nematode, Bursaphelenchus xylophilus, in Minnesota and Wisconsin: Insect associates and transmission studies. Canadian Journal of Forest Research 13, 1068–1076 (1983).
Smith, S. G. Chromosome numbers of Coleoptera. Heredity 7, 31–48 (1953).
Deshpande, A. S. et al. Identifying synergistic high-order 3D chromatin conformations from genome-scale nanopore concatemer sequencing. Nature Biotechnology 40, 1488–1499 (2022).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature Communications 11, 1432 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175 (2021).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: Allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19, 460 (2018).
Laetsch, D. R. & Blaxter, M. L. BlobTools: Interrogation of genome assemblies. F1000Research 6, 1287–1287 (2017).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Systems 3, 99–101 (2016).
Alonge, M. et al. Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome Biology 23, 1–19 (2022).
Chen, Y., Zhang, Y., Wang, A. Y., Gao, M. & Chong, Z. Accurate long-read de novo assembly evaluation with Inspector. Genome Biology 22, 321 (2021).
Kerpedjiev, P. et al. HiGlass: Web-based visual exploration and analysis of genome interaction maps. Genome Biology 19, 125 (2018).
Uliano-Silva, M. et al. MitoHiFi: A python pipeline for mitochondrial genome assembly from PacBio high fidelity reads. BMC Bioinformatics 24, 288 (2023).
Allio, R. et al. MitoFinder: Efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Molecular Ecology Resources 20, 892–905 (2020).
Baril, T., Galbraith, J. & Hayward, A. Earl Grey: A fully automated user-friendly transposable element annotation and analysis pipeline. Molecular Biology and Evolution 41, 1–18 (2024).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. Retrieved from https://www.repeatmasker.org (2023).
Storer, J., Hubley, R., Rosen, J., Wheeler, T. J. & Smit, A. F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mobile DNA 12, 1–14 (2021).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences of the United States of America 117, 9451–9457 (2020).
Platt, R. N., Blanco-Berdugo, L. & Ray, D. A. Accurate transposable element annotation is vital when analyzing new genome assemblies. Genome Biology and Evolution 8, 403–410 (2016).
Xu, Z. & Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–W268 (2007).
Gabriel, L. et al. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Research 34, 769–777 (2024).
Bushmanova, E., Antipov, D., Lapidus, A. & Prjibelski, A. D. RnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 8, 1–13 (2019).
Haas, B. J. TransDecoder (version 5.7.0) Retrieved from https://github.com/TransDecoder/TransDecoder (2023).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O. & Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research 33, 6494–6506 (2005).
Brůna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP+: Eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics 2, 1–14 (2020).
Gabriel, L., Hoff, K. J., Brůna, T., Borodovsky, M. & Stanke, M. TSEBRA: Transcript selector for BRAKER. BMC Bioinformatics 22, 1–12 (2021).
Caballero, M. & Wegrzyn, J. gFACs: Gene filtering, analysis, and conversion to unify genome annotations across alignment and gene prediction frameworks. Genomics, Proteomics & Bioinformatics 17, 305–310 (2019).
Cantalapiedra, C. P., Hernnandez-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Molecular Biology and Evolution 38, 5825–5829 (2021).
Altschul, S. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Research 25, 3389–3402 (1997).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nature Methods 18, 366–368 (2021).
Schultz, D. T. et al. Ancient gene linkages support ctenophores as sister to other animals. Nature 618, 110–117 (2023).
Emms, D. M. & Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biology 20, 1–14 (2019).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – Approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution 30, 772–780 (2013).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution 37, 1530–1534 (2020).
Zhang, C., Rabiee, M., Sayyari, E. & Mirarab, S. ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19, 15–30 (2018).
Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution 24, 1586–1591 (2007).
Wang, B. et al. The earliest known longhorn beetle (Cerambycidae: Prioninae) and implications for the early evolution of Chrysomeloidea. Journal of Systematic Palaeontology 12, 565–574 (2013).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR34444379 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR34444378 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR34444377 (2025).
Kim, S. & Farrell, B. D. Chromosome-level genome assembly of the white-spotted sawyer beetle Monochamus scutellatus (Coleoptera: Cerambycidae). GenBank https://identifiers.org/ncbi/insdc.gca:GCA_052862855.1 (2025).
Kim, S. & Farrell, B. D. Chromosome-level genome assembly of the white-spotted sawyer Monochamus scutellatus (Coleoptera: Cerambycidae). figshare https://doi.org/10.6084/m9.figshare.29575361 (2025).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Kriventseva, E. V. et al. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Research 47, D807–D811 (2019).
Lewin, H. A. et al. Earth BioGenome Project: Sequencing life for the future of life. Proceedings of the National Academy of Sciences of the United States of America 115, 4325–4333 (2018).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Acknowledgements
Computational analyses were performed on the FASRC Cannon cluster supported by the FAS Division of Science Research Computing Group, Harvard University. This work was supported by the Graduate Research Fund of the Department of Organismic and Evolutionary Biology and the Dean’s Competitive Fund for Promising Scholarship at Harvard University; and grants from the National Institute of Biological Resources, the Ministry of Environment (MOE), Republic of Korea (No. NIBR202405101) and the National Research Foundation of Korea, funded by the Ministry of Education, Science and Technology (MEST) (No. 2019R1A6A1A10073437). Fieldwork was supported by the Putnam Expedition Grant, and publication costs were covered by the Wetmore Colles Fund of the Museum of Comparative Zoology, Harvard University.
Author information
Authors and Affiliations
Contributions
Sangil Kim: Conceptualization; data curation; formal analysis; funding acquisition; investigation; methodology; project administration; resources; validation; visualization; writing – original draft; writing – review and editing. Brian D. Farrell: Conceptualization; data curation; funding acquisition; investigation; methodology; project administration; resources; supervision; validation; writing – review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, S., Farrell, B.D. Chromosome-level genome assembly of the white-spotted sawyer Monochamus scutellatus (Coleoptera: Cerambycidae). Sci Data 12, 2009 (2025). https://doi.org/10.1038/s41597-025-06258-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06258-0
