
Abstract
Swertia przewalskii Pissjauk. belonging to Swertia (subtribe Swertiinae, Gentianaceae), is one of the original plant sources of the Tibetan medicine “Zangyinchen” has been included in the initial list of plant species with extremely small populations in Qinghai province. To gain deeper insights into the threats to S. przewalskii survival, we assembled a high-quality, chromosome-level genome using a combination of Illumina short-read, PacBio HiFi long-read, and Hi-C scaffolding technologies. The assembled genome has a size of ~2027.49 Mb, and demonstrates excellent continuity, consistency, and completeness (contig N50 = 2,569,777, mapping rate = 98.87%, coverage = 99.29%, BUSCO = 96.9%). The assembled sequences could be anchored to twelve chromosomes. Additionally, genome annotation revealed 35,701 protein-coding genes, 289 microRNAs (miRNAs), 1,039 transfer RNAs (tRNAs), 3,681 ribosomal RNAs (rRNAs). 32,775 genes were functionally annotated using several databases, including NR (31,587, 88.48%), GO (23,883, 66.90%), KOG (663, 1.86%), and Pfam (22,995, 64.41%), KEGG (11,964, 33.51%). This study reports a high-quality, chromosome-level genome assembly of S. przewalskii, providing a foundational resource for its conservation biology.
Similar content being viewed by others
Background & Summary
While the decline in global biodiversity has been a long-standing concern, the compounding effects of habitat destruction and climate change have now made it a pressing crisis1. According to the State of the World’s Plants and Fungi 2020 report released by the Royal Botanic Gardens, Kew in the United Kingdom, over 30% of known plant species worldwide are currently threatened with extinction. In China, this trend is reflected in the nearly 5,000 plant species that have been classified as endangered2. Therefore, to provide effective and scientifically-grounded protection for these endangered species, researchers have proposed the concept of Plant Species with Extremely Small Populations (PSESP)3. The concept characterized by narrow geographic distribution, persistent intrinsic and extrinsic constraints, visible population degradation, and ongoing demographic decline, has attracted considerable attention, rapidly becoming a central focus of biodiversity conservation in China4,5,6. Moreover, advancements in whole-genome sequencing technologies have greatly facilitated related conservation work, enabling more in-depth investigations into the mechanisms underlying endangerment in many PSESP species7,8,9,10.
As a species endemic to the Qinghai-Tibetan Plateau (QTP), the S. przewalskii is primarily found in Qilian County, Qinghai Province11. It belongs to the genus Swertia (subtribe Swertiinae, Gentianaceae), is a perennial herb, and one of the original source plants of Tibetan medicine “Zangyinchen”11. It contains a rich array of chemical constituents (such as loganic acid, sweroside, and gentiopicroside) and exhibits significant anti-inflammatory effects12. The species was included in the initial list of plant species with extremely small populations in Qinghai province due to its narrow distribution range, and the extremely limited number of wild populations13. In recent years, research on the taxonomy, phylogeny, and population genetics of S. przewalskii has been conducted. However, studies focusing on its conservation genetics remain scarce14,15,16. Therefore, it is crucial to investigate the mechanisms underlying endangerment of S. przewalskii.
Herein, we reported the first chromosome-level genome assembly of S. przewalskii, achieved through a combination of three sequencing strategies: next-generation sequencing, third-generation SMRT (Single Molecule Real-Time) sequencing, and high-throughput chromosome conformation capture (Hi-C) sequencing. This high-quality genome provides a valuable resource for analyzing the genetic factors underlying the endangerment of S. przewalskii. Building on this, our study further deepens the understanding of its genetic background and offers scientific evidence to support the conservation of this endangered species.
Methods
Plant materials
Specimens of the study species were collected and their morphological traits were examined in details. The stem was erect and yellow-green, with black-brown withered leaf petioles remaining at the base. Each plant bore 1-2 pairs of basal leaves, with blades ranging from oval to ovate-elliptic or spoon-shaped. The inflorescence formed a narrow umbel comprising 3–9 flowers. Detailed observations of the flowers revealed that the calyx extended to approximately two-thirds the length of the corolla, which was yellow-green with a blue center that gradually turned brown with age. The anthers were blue and either oval or narrow rectangular, while the ovary surface often displayed transverse wrinkles17 (Fig. 1A).

Ecological map of S. przewalskii, and the genome assembly and annotation of S. przewalskii. (A) Photograph of S. przewalskii, showing its natural habitat, and morphological features. (B) The process pipeline of S. przewalskii genome assembly and annotation.
The fresh root, stem, leaf, and flower tissues (37° 36′ 0.00″N, 100° 42′ 0.00″E, at an elevation of 3486 m) were harvested from a mature individual of S. przewalskii. To ensure optimal preservation of the collected samples and to prevent RNA degradation, the plant tissues were treated in the field by rinsing twice with ultrapure water and once with a 75% ethanol, transferred into Eppendorf (EP) tubes, and immediately frozen in liquid nitrogen. Upon return to the laboratory, the samples were subsequently stored at −80 °C until further use.
Library preparation and sequencing
For next-generation sequencing, the genomic DNA was extracted from S. przewalskii employing the CTAB (Hexadecyl trimethyl ammonium Bromide) protocol, which is widely used for plant DNA isolation due to its efficiency in removing polysaccharides and polyphenols18. Genomic DNA (gDNA) quality and quantity were carefully assessed prior to library construction. The integrity and degree of fragmentation of DNA were evaluated using agarose gel electrophoresis, while DNA purity and concentration were measured using a NanoDropTM One UV-Vis19 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and a Qubit 3.0 fluorometer20 (Life Technologies, Carlsbad, CA, USA), respectively. High-quality gDNA fragments of approximately 150 bp were then selected to construct sequencing libraries, ensuring optimal fragment size for downstream analyses. The DNA libraries were initially quantified using the Qubit 2.0 fluorometer21, and after appropriate dilution, the fragment size quality was evaluated with an Agilent 2100 Bioanalyzer22. Finally, the effective library concentration was determined by quantitative PCR (Q-PCR)23 to ensure suitability for downstream sequencing applications. After passing these quality control step, next-generation sequencing was performed on the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA).
For third-generation SMRT (Single Molecule Real Time) sequencing, genomic DNA was first extracted using the same procedure as for next-generation sequencing. The high-quality genomic DNA was then fragmented to appropriated sizes, followed by damage repair, adapter ligation to construct the sequencing library. After selecting the desired fragments and quantifying the library, a PCR-free SMRTbell library was prepared. Finally, the library templates were combined with the sequencing enzyme complexes and loaded onto the PacBio Revio platform for high-throughput sequencing.
For Hi-C sequencing, genomic DNA was extracted from fresh leaves and fixed with formaldehyde, followed by digestion with the restriction endonuclease DPNII. The quality of the DNA was assessed according to standard next-generation sequencing protocols, including evaluation of DNA fragment size, degree of degradation, and measurement of purity and concentration. Hi-C libraries were then constructed through a series of steps, including cell crosslinking, endonuclease digestion, end repair, cyclization, DNA purification, and capture. The resulting libraries were subjected to high-throughput sequencing on the Illumina platform (Fig. 1B).
Data filtering and genomic assembly
Next-generation sequencing data were first subjected to quality control to remove low-quality reads and adapter sequences using Fastp v 0.21.024 with default parameters, yielding high-quality clean reads for downstream analyses. For third-generation SMRT sequencing, raw polymerase reads were processed to retain only the Subreads corresponding to the insert sequences. Adaptors sequences and low-quality reads were subsequently removed using SMRTlink v12.0 (https://www.pacb.com/support/software-downloads), producing high-quality reads suitable for accurate genome assembly. For Hi-C sequencing, raw data were subjected to rigorous quality control, during which low-quality sequences were removed to obtain clean reads. To obtain an initial insight into the genomic characteristics, genome size and heterozygosity were estimated utilizing a K-mer-based strategy. Specifically, genome size was assessed with GCE v1.0.025. In parallel, Jellyfish v2.2.1026 was used to calculate the K-mer frequency-depth distribution, which provided an additional basis for genome size estimation. Following quality control and filtering of raw sequencing data, next-generation short reads and HiFi reads were employed for genome assembly of S. przewalskii. Short reads provided high base accuracy, facilitating error correction of sequencing, while HiFi reads span repetitive and complex regions, enabling continuous contig construction. In general, hifiasm v0.16.1 (https://github.com/chhylp123/hifiasm) assembled the genome using an overlap-layout-consensus (OLC) algorithm. HiFi reads were first aligned using all-versus-all method to detect overlaps, and three rounds of self-correction were performed to reduce sequencing errors. This process was iteratively repeated, allowing overlaps to be refined and a string graph to be constructed for contig-level genome assembly. The resulting contigs were then scaffolded to chromosome-level assemblies using Hi-C interaction data. Hi-C valid interaction pairs were employed to cluster contigs into chromosome groups and determine their linear order and orientation using ALLHIC v0.9.827, with manual refinement performed based on Hi-C contact maps. Interaction data were converted into binary format using 3D-DNA v 18041928 and jucier v1.629. The Manual refinement of contig order and orientation was performed with Juciebox v 1.11.0830. Redundant sequences were identified and removed with Purge_dups v 1.2.5 (https://github.com/dfguan/purge_dups), further optimizing the genome assembly. To generate a chromosome-level assembly, gaps were filled using placeholder sequences of 100 Ns. Finally, HiCExplorer v 3.631 was used to visualize chromosomal interactions via Hi-C heatmaps. To comprehensively evaluate the quality of the genome assembly, we calculated its continuity, consistency, and completeness. The contig N50 was used to quantify assembly continuity. Clean reads from next-generation sequencing were aligned to the reference genome using BWA-MEM v0.7.1732, providing mapping rate and coverage as indicators of assembly consistency. Finally, genome completeness was evaluated with BUSCO v5.3.0 v 5.3.033 (parameter setting: Busco -m prot -c 16).
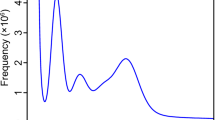
A comprehensive sequencing effort was undertaken using three sequencing strategies, yielding a total of 136.54 GB of Illumina short-read data, 81.71 GB of PacBio HiFi long-read data, and 268 GB of Hi-C data. Following quality control, 909,483,996 clean reads were retained from the Illumina dataset out of 910,278,082 raw reads, with Q20 and Q30 scores reaching 98.38% and 95.42%, respectively, reflecting high sequencing quality. The PacBio HiFi dataset comprised 5,057,384 reads with an average length of 16,157 bp. For the Hi-C data, 1,791,494,328 reads were retained after filtering, and both Q20 and Q30 scores exceeded 90%, demonstrating superior sequencing quality (Table 1), Collectively, these datasets provide a robust foundation for high-quality genome assembly. The 19-mer frequency analysis yielded a genome size estimate of ~2027.49 Mb for S. przewalskii, with the distribution peak suggesting low heterozygosity (1.14%, ~ 66.71 X) and a largely homozygous genome (Fig. 2A). The genome assembly exhibited high continuity, with a contig N50 of 2,569,777 bp. Mapping Illumina clean reads to the reference genome revealed a coverage and alignment rate exceeding 90%, further supporting assembly accuracy. Assessment of gene content using BUSCO identified X complete orthologs, corresponding to over 95% completeness (Table 2), indicating that the assembled genome is both consistent and complete. The sequences were successfully anchored to twelve chromosomes, and Hi-C interaction heatmaps confirmed the integrity and high quality of the chromosome-level assembly (Fig. 2B,C).

The assembly and characteristics of chromosome-level genomes of S. przewalskii. (A) The distribution of depth and frequence for Kmer = 19. (B) The Hic heatmap for interaction of twelve chromosomes, which indicates that a better genome anchoring. C The synteny and distribution of chromosome-level genomes of S. przewalskii. (I) synteny of gene for S. przewalskii genome. (II) GC content. (III) the density of repeat sequences. (IV) gene density (V) the twelve chromosomes.
Genome annotations
The genome annotation of S. przewalskii encompassed the identification of repetitive sequences, protein-coding genes and its models, and non-coding RNAs (ncRNA). Repetitive sequences were annotated using a combination of Ab initio and Repbase-based approaches to maximize sensitivity and accuracy. For Ab initio prediction, RepeatModeler v open-1.0.1134 and RepeatMasker v open-4.0.935 were employed with default parameters to construct de novo repeat library and identify repeat elements. In parallel, Repbase-based prediction was conducted using RepeatModeler v 2.0.434 with the parameter of -database mydb -threads 16, referencing the Repbase database to identify both DNA- and protein-level repeat sequences. To eliminate redundancy, LTR retriever v 2.9.036 was utilized to obtain non-redundant long terminal repeat sequences from the combinated results of the Ab initio and Repbase-based searches. These two De novo sequence sets were subsequently integrated to form a comprehensive De novo repeat library. Repeated sequences were then aligned and predicted by integrating the De novo and Repbase databases, generating a consolidated annotation of repetitive elements. Transposable element sequences were further predicted using RepeatProteinMask v 4.1.5 (https://github.com/Dfam-consortium/RepeatMasker), and all repeat annotation results were merged with redundancies removed to produce the final high-confidence set of transposable elements. Additionally, tandem repeats were identified utilizing TRF v 4.0937 and MISA v 2.138, completing the characterization of repetitive sequences within the genome. In this study, repetitive sequences were annotated using a combined strategy of ab initio prediction and Repbase-based homology searches. Allowing for comprehensive identification of various repeat types, long terminal repeats (LTRs) were the most abundant, accounting for 74.73% of the genome, followed by DNA elements (6.93%), LINEs (4.77%), SINEs (0.26%), and Unknown (2.13%) (Fig. 3A,B). Tandem repeats comprised a total of 4.963% of the genome, including minisatellite (repeat units of 10–99 bp, 2.886%), satellite (repeat units ≥ 100 bp, 1.823%), and microsatellite (repeat units of 1–9 bp, 0.823%) (Table S1). These results provide a detailed overview of the repetitive landscape of the S. przewalskii genome, highlighting the predominance of LTRs and the contribution of tandem repeats to genome structure.

The repeat sequences analysis in S. przewalskii genome. (A) the divergence rate and percentage of genome in the four transposable elements. (B) the statistical analysis of all transposable elements in S. przewalskii genome. X-axis: the sequence divergence rate of each TE, Y-axis: the percentage of S. przewalskii genome for each TE.
To facilitate comprehensive genome annotation of S. przewalskii, it was essential to obtain detailed information on gene distribution and structure. We employed an integrative strategy combining transcriptome-based prediction, homology prediction, and De novo prediction to maximize accuracy and completeness. For transcriptome-based prediction, both Illumina dataset and PacBio dataset were used. Raw Illumina reads were quality-filtered using fastp v 0.23.224, Hi-C reads with fastp v 0.21.024 (https://github.com/wdecoster/nanofilt), and PacBio reads with isoseq 3 v 3.9.0 (https://github.com/ylipacbio/IsoSeq3). Genome and reads alignments were carried out with Hisat2 v 2.2.139 for Illumina data, HICUP v 0.8.040 for Hi-C data, and Pbmm2 v 1.10.0 (https://github.com/PacificBiosciences/pbmm2) for PacBio data, enabling accurate mapping of reads to the reference genome. Coding sequences were predicted using TransDecoder v 5.7.0 (https://github.com/TransDecoder/TransDecoder), while homologous protein sequences were aligned to the genome with tblastn v 2.13.041, and transcript structures were further refined using Exonerate v 2.4.042. De novo gene prediction was conducted by Augustus v 3.5.043, trained on a specific parametric model. Additionally, gene set derived from the three strategies were integrated using Maker v 3.01.0344, generating a high-quality, non-redundant annotation. Through this integrative approach, we predicted a total of 35,701 protein-coding genes. The average coding sequence (CDS) length per gene was 1,089.01 bp, with each gene containing an average of 4.97 exons (Table 3). In total, 177,330 exons were identified with an average length of 306.64 bp, alongside 141,629 introns (average length: 1267.94 bp, total length: 179,577,541 bp) (Table 4). This comprehensive annotation provides a robust foundation for downstream functional and comparative genomic analyses. We further annotated multiple non-coding RNA, including 289 of miRNAs (0.0016%), 1,039 of tRNAs (0.0036%), 3,681 of rRNAs (0.2243%), and 9,244 of snRNAs (0.0473%), to achieve a more comprehensive genome annotation (Table 5). Additionally, the genome characteristics of S. przewalskii was visualized using R package “circlize” with a 50 kb sliding window, providing an intuitive overview of gene density, repeat content, and other genomic elements across chromosomes. Assessment of genome completeness revealed that over 95% of the completed BUSCOs were present, reflecting a high-quality and complete genome annotation.
To comprehensively characterize the functional landscape of protein-coding genes in S. przewalskii, we employed a dual annotation strategy, integrating both sequence similarity and motif/domain-based approaches. First, predicted protein sequences were compared against multiple databases, including universal protein45, non-Redundant Protein Database (NR)46, cluster of orthologous groups of proteins (COG)47, and eukaryotic orthologous groups (KOG), using diamond v 2.1.8, allowing rapid and sensitive identification of homologous proteins. Second, motif and domain-based annotations were conducted with InterProScan v 5.55-88.048, querying a suite of databases, including CDD, Gene3D, Hamap, Phobius, Pirsf, Prosite, Sfld, Superfamily, Tigrfam, Tmhmm and others. Conserved domains and sequence motifs were further identified using hmmscan v 3.3.249. Functional insights were then expanded through metabolic pathway assignment based on KOfam50 profiles within the Kyoto Encyclopedia of Genes and Genomes (KEGG)51 database. In parallel, non-coding RNA annotations were conducted to capture regulatory and structural elements. Transfer RNAs (tRNAs) were identified with tRNAscan-SE v 2.0.1252, ribosomal RNAs (rRNAs) with RNAmmer v 1.253, and ncRNA sequences with INFERNAL v 1.1.454, against the RNA family (Rfam, version: 14)55 database. Collectively, a total of 32,775 genes (91.80%) were functionally annotated across multiple databases, including NR (31,587; 88.48%), GO (23,883; 66.90%), KOG (663; 1.86%), and Pfam (22,995; 64.41%), KEGG (11,964; 33.51%) (Fig. 4, Table 6), providing a robust and multidimensional view of gene functions, conserved domains, and metabolic pathways in the S. przewalskii genome.

The analysis of gene function annotation of S. przewalskii genome. The gray circles on the vertical bars indicate overlapping annotations, such as genes identified by one or more databases. The total number of genes annotated to each database is shown by the horizontal bars.
Data Records
All the sequencing data have been deposited in the National Genomics Data Center (NGDC), Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation56,57 (Project accession number: PRJCA032197). Illumina short-reads, PacBio HiFi long-reads, Hi-C reads have been deposited in the Genome Sequence Archive (GSA) in NGDC under the accession number CRR137065758, CRR137065859, CRR137065960 that is publicly accessible at https://download.cncb.ac.cn/gsa2/CRA020335. S. przewalskii chromosome-level genome assembly and gene annotation files have been deposited in the Genome WareHouse (GWH) in NGDC under the accession number is GWHFIGX00000000.161 that is publicly accessible at https://ngdc.cncb.ac.cn/gwh. The genomic Illumina, PacBio, and Hi-C sequencing raw data were also deposited in the European Nucleotide Archive (ENA) at EMBL - EBI62 with accession number ERR1569609163, ERR1569632164, ERR1572902465 (Study accession number: PRJEB100542; Sample accession number: ERS26986347). S. przewalskii chromosome-level genome assembly and annotated files were deposited in the European Nucleotide Archive (ENA) at EMBL - EBI under the accession number is GCA_97701221566.
Technical Validation
In the present study, we utilized a NanoDropTM One UV-V spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and Qubit 3.0 fluorometer (Life Technologies, Carlsbad, CA, USA) to measure the DNA purity and concentration, respectively. After library preparation, sequencing was performed using three sequencing strategies. From these datasets, we filtered raw data, and retain high-quality clean data. We then utilized the19-mer frequency analysis to estimate genome size for S. przewalskii. Additionally, we used hifiasm and Purge_dups to discard genome redundancy sequence, producing a draft genome. We further anchored the genome sequences to twelve chromosomes, covering 95.02% of the genome, and the Hi-C interaction heatmap confirmed the accuracy and continuity of chromosome-level scaffolding. This comprehensive genome assembly provides a valuable resource, significantly enhancing our understanding of S. przewalskii and establishing a solid foundation for future functional and evolutionary studies.
Data overview
Sequencing was performed, yielding a total of 136.54 GB of Illumina data, 81.71 GB of PacBio HiFi data, and 268 GB of Hi-C data using three sequencing strategies. From these datasets, we obtained 909,483,996 clean reads from Illumina dataset out of 910,278,082 raw reads. Q20 and Q30 score was 98.38% and 95.42%, respectively, indicating high sequencing quality. Additionally, 5,057,384 reads were generated for PacBio HiFi data, with an average read length of 16,157 bp. For Hi-C data, 1,791,494,328 reads were retained after filtering, and both Q20 and Q30 scores exceeded 90%, demonstrating superior sequencing quality for Hi-C strategy.
We further evaluated the quality of the S. przewalskii genome assembly in terms of contiguity, completeness, and consistency. The assembly exhibited a contig N50 of 2,569,777, with 96.9% of the BUSCOs as complete, a mapping rate of 98.87%, and a coverage rate of 99.29%, indicating a well-assembled genome. The completeness of genome annotation was also assessed, exceeding 90%, reflecting a high-quality annotation. S. przewalskii chromosome-level genome comprised 35,701 protein coding genes, 1,039 tRNAs, 3,681 rRNAs, and 289 miRNAs. A total of 32,775 genes (91.80%) were functionally annotated across multiple databases, including NR (31,587; 88.48%), GO (23,883; 66.90%), KOG (663; 1.86%), and Pfam (22,995; 64.41%), KEGG (11,964; 33.51%), providing a comprehensive view of gene content, structure, and functional potential in S. przewalskii.
Data availability
The sequencing raw data generated in this study are available at ENA https://www.ebi.ac.uk/ena/browser/view/PRJEB100542 and CNCB-GSA https://ngdc.cncb.ac.cn/gsa/browse/CRA020335. The genome assembly files generated in this study are available at ENA http://www.ebi.ac.uk/ena/browser/view/GCA_977012215 and Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/88049/show.
Code availability
All raw sequencing data were processed and quality-checked to ensure high fidelity for downstream analyses. Illumina raw data and PacBio raw data were filtered by FastQC v0.20.1 and Pbmm2 v1.10.0, respectively. All software and scripts used in this study are publicly accessible and freely available, with parameters set according to the respective manuals, including filters --low_complexity_filter, --adapter_sequence, seqkit rmdup). ALLHiC software version 0.9.8 for genome assembly is used as a free, publicly available at GitHub: https://github.com/tangerzhang/ALLHiC. Hi-C based scaffolding and contact map generation were achieved using 3D-DNA version 180922 (https://github.com/aidenlab/3d-dna) and Juicer version 1.6 (https://github.com/aidenlab/juicer), respectively. Denovo repeated sequence prediction was performed using RepeatModeler version 2.0.4 (https://www.repeatmasker.org/RepeatModeler/, parameters setting: -database mydb -threads 16). LTR sequences prediction was carried out utilizing LTR_FINDER (https://github.com/oushujun/LTR_FINDER_parallel, parameters setting: -threads 16 -harvest_out -size 1000000 -time 300), and LTRharvest version 1.62 (http://genometools.org/pub/binary_distributions, parameters setting: -minlenltr 100 -maxlenltr 7000 -mintsd 4 -maxtsd 6 -motif TGCA -motifmis 1 -similar 85 -vic 10 -seed 20 -seqids yes). Transposable elements prediction was performed using RepeatProteinMask version 4.1.5 (https://www.repeatmasker.org/RepeatProteinMask.html, parameters setting: -noLowSimple -pvalue 0.0001). Tandem repeats prediction was carried out utilizing TRF version 4.09 (https://tandem.bu.edu/trf/trf.html, parameters setting: 2 7 7 80 10 50 2000 -d -h) and MISA version 2.1 (https://webblast.ipk-gatersleben.de/misa/). Transcriptome-based gene prediction was performed using Tama version 1.0 (https://github.com/GenomeRIK/tama, parameters setting: -f filelist.txt -p merge), stringtie version 2.2.1 (https://ccb.jhu.edu/software/stringtie/, parameters setting: -p 16 -R -L), and TransDecoder version 5.7.0 (https://github.com/TransDecoder/TransDecoder, parameters setting: TransDecoder. Predict -retain_long_orfs_length 150, TransDecoder.LongOrfs -m 50). Homology-based gene prediction was carried out using Exonerate version 2.4.0 (https://github.com/nathanweeks/exonerate, parameters setting: -model protein2genome -showtargetgff 1), and tblast version 2.13.0 (default parameters). De novo gene prediction was performed using Augustus version 3.5.0 (https://github.com/Gaius-Augustus/Augustus, parameters setting: -uniqueGeneId = true -noInFrameStop = true -gff3 = on -strand = both), and glimmerhmm version 3.0.4 (http://ccb.jhu.edu/software/glimmerhmm/, parameters setting: -f -g). Evaluating the completeness of genome was performed utilizing BUSCO version 5.4.7 (https://busco.ezlab.org/, parameters setting: busco -m prot -c 16). Gene function annotation based on sequence similarity searching was carried out using diamond version 2.1.8 (https://github.com/bbuchfink/diamond, default parameters). Gene function annotation based on motif similarity searching was performed utilizing InterProScan version 5.55–88.0 (https://github.com/ebi-pf-team/interproscan, default parameters), and hmmscan version 3.3.2 (http://hmmer.org/download.html, parameters setting: -cpu 16 -E 1e-5). Non-coding RNAs (ncRNAs) were predicted using tRNAscan-SE version 2.0.12 (https://github.com/UCSC-LoweLab/tRNAscan-SE, parameters setting: -E -j tRNA.gff -o tRNA.result -f tRNA.struct -thread 16), RNAmmer version 1.2 (https://services.healthtech.dtu.dk/services/RNAmmer-1.2/, parameters setting: -S euk -m tsu, lsu, ssu), and INFERNAL version 1.1.4 (http://eddylab.org/infernal/, parameters setting: -Z (genome_length2CMmumber/10) -cut_ga -rfam -nohmmonly -fmt 2).
References
Huang, J. H. et al. Identifying hotspots of endemic woody seed plant diversity in China. Divers. Distrib. 18, 673–688 (2011).
Xu, Y. & Zang, R. G. Conservation of rare and endangered plant species in China. IScience 26, 106008 (2023).
Sun, W. B. Conservation of plant species with extremely small populations in Yunnan - practice and exploration. Yunnan Science and Technology Press, Kunming, China (2013).
Sun, W. B. Words from the Guest Editor-in-Chief. Plant Divers 38, 207–208 (2016).
Sun, W. B., Ma, Y. P. & Blackmore, S. How a new conservation action concept has accelerated plant conservation in China. Trends Plant Sci 24, 4–6 (2019a).
Sun, W. B., Yang, J., Dao, Z. L. Study and conservation of plant species with extremely small populations (PSESP) in Yunnan province, China. Science Press, Beijing, China (2019b).
Yang, T. et al. The chromosome-level genome assembly and genes involved in biosynthesis of nervonic acid of Malania oleifera. Sci. Data. 10, 298 (2023).
Shi, C. Y. et al. A high-quality chromosome-level genome assembly of the endangered tree Kmeria septentrionalis. Sci. Data. 11, 775 (2024).
Song, Y. et al. Chromosome level genome assembly of endangered medicinal plant Anisodus tanguticus. Sci. Data. 11, 161 (2024).
Tan, W., Zhou, P. Y. & Huang, X. Haplotype-resolved genome of Prunus zhengheensis provides insight into its evolution and low temperature adaptation in apricot. Hort. Res. 11, 2662–6810 (2024).
Liu, J. Z., Chen, Z. D. & Lu, A. M. A preliminary analysis of the phylogeny of the Swertiinae (Gentianaceae) based on ITS data. Isr. J. Plant Sci. 49, 301–308 (2001).
Liu, H. G. et al. Genetic relationship and resources evaluation of medicinal plants in Swertia L. Chin. Herb. Med. 42, 1646–1650 (2011).
Ji, L. J. et al. Study on chemical compositions of four Gentiana plantsfrom qinghai-xizang plateau. Acta Biol. Plateau Sin. 11, 113–118 (1992).
Zhang, D. F. et al. Patterns of genetic variation in Swertia przewalskii, an endangered endemic species of the Qinghai-Tibet Plateau. Biochem. Genet. 45, 33–50 (2007).
Duan, Y. W., Liu, J. Q. Pollinator shift and reproductive performance of the Qinghai-Tibetan Plateau endemic and endangered Swertia przewalskii (Gentianaceae). In: Hawksworth, D.L., Bull, A.T. (eds) Plant Conservation and Biodiversity. Topics in Biodivers. Conserv. 6. S. przewalskiiinger, Dordrecht (2006).
Cao, Q. et al. Plastome structure, phylogenomics and evolution of plastid genes in Swertia (Gentianaceae) in the Qinghai-Tibetan Plateau. BMC Plant Biol. 22, 195 (2022).
Ho, T. N., & Pringle, J. S. Gentianaceae. In Z. Y. Wu & P. H. Raven (Eds.), Flora of China (Vol. 16, pp. 1–139). Science Press & Missouri Botanical Garden Press. (1995).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15 (1987).
Thermo Fisher Scientific. (n.d.). NanoDrop One UV-V Spectrophotometer. Thermo Fisher Scientific. Retrieved from https://www.thermofisher.com.
Thermo Fisher Scientific. (n.d.). Qubit 3.0 Fluorometer. Thermo Fisher Scientific. Retrieved from https://www.thermofisher.com.
Thermo Fisher Scientific. (n.d.). Qubit 2.0 Fluorometer. Thermo Fisher Scientific. Retrieved from https://www.thermofisher.com.
Agilent Technologies. (n.d.). Agilent 2100 Bioanalyzer. Agilent Technologies. Retrieved from https://www.agilent.com.
Heid, C. A. et al. Real-time quantitative PCR. Genome Research 6(10), 986–994, https://doi.org/10.1101/gr.6.10.986 (1996).
Chen, S. F. et al. Fastp: an ultra-fast all-in-one FASTQ preproces-sor. Bioinformatics. 34, 884–890 (2018).
Liu, B. H. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quant. Biol. 35, 62–67 (2013).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764–770 (2011).
Zhang, X. et al. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on hi-c data. Nat. Plants. 5, 833–845 (2019).
Dudchenko, O. et al. De novo assembly of the aedes aegypti genome using hi-c yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution hi-c experiments. Cell Systems. 3, 95–98 (2016b).
Durand, N. C. et al. Juicebox provides a visualization system for hi-c contact maps with unlimited zoom. Cel. systems. 3, 99–101 (2016a).
Wolff, J. et al. Galaxy HiCExplorer 3: A web server for reproducible hi-c, capture hi-c and single-cell hi-c data analysis, quality control and visualization. Nucleic Acids Res, Oxford University Press. 48, W177–W184 (2020).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv (2013).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. 62, 227–245 (2019).
Smit, A., Hubley, R. & Green, P. RepeatModeler open-1.0 (2008–2015) Seattle, USA: Institute for Systems Biology. Available from: https://www.repeatmasker.org/, Last Accessed May, 1, 2018 (2015).
Smith, A., Hubley, R. & Green, P. RepeatMasker open-4.0, RepeatMasker Open-4.0. (2013).
Ou, S. & Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res, Oxford University Press. 27, 573–580 (1999).
Beier, S. et al. MISA-web: A web server for microsatellite prediction. Bioinformatics, Oxford University Press. 33, 2583–2585 (2017).
Kim, D. et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Wingett, S. et al. HiCUP: Pipeline for mapping and processing hi-c data, F1000Research, Faculty of 1000 Ltd, 4 (2015).
Camacho, C. et al. BLAST+: Architecture and applications. BMC Bioinform. 10, 1–9 (2009).
Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 6, 1–11 (2005).
Stanke, M. et al. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 24, 637–644 (2008).
Holt, C. & Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 12, 1–14 (2011).
Apweiler, R. et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 32, D115–D119 (2004).
Deng, Y. et al. Integrated nr database in protein annotation system and its localization. Comput. Eng. 32, 71–72 (2006).
Tatusov, R. L. et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 4, 1–14 (2003).
Blum, M. et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 49, D344–D354 (2021).
Mistry, J. et al. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41, e121–e121 (2013).
Aramaki, T. et al. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 36, 2251–2252 (2020).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Chan, P. P. et al. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. BioRxiv. 614032 (2021).
Lagesen, K. et al. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29, 2933–2935 (2013).
Kalvari, I. et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 49, D192–D200 (2021).
Chen, M. et al. Genome Warehouse: A Public Repository Housing Genome-scale Data. Genom. Proteom. Bioinfo. 19, 584–589 (2021).
Database Resources of the National Genomics Data Center. China National Center for Bioinformation in 2022. Nucleic Acids Res. 50, D27–D38 (2022).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA020335/CRR1370657 (2024).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA020335/CRR1370658 (2024).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA020335/CRR1370659 (2024).
NGDC Genome WareHouse https://ngdc.cncb.ac.cn/gwh/Assembly/88049/show (2024).
European Nucleotide Archive (ENA) https://www.ebi.ac.uk/ena.
European Nucleotide Archive (ENA); ERR15696091 https://identifiers.org/insdc.sra:ERR15696091 (2025).
European Nucleotide Archive (ENA); ERR15696321 https://identifiers.org/insdc.sra:ERR15696321 (2025).
European Nucleotide Archive (ENA) ERR15729024 https://identifiers.org/insdc.sra:ERR15729024 (2025).
European Nucleotide Archive (ENA); GCA_977012215 https://identifiers.org/insdc.gca:GCA_977012215 (2025).
Acknowledgements
This research was partially supported by the Qinghai Provincial Science and Technology Major Project (2023-SF-A5), the Xining Science and Technology Major Project (2023-Z-13), the CAS-Qinghai on Sanjiangyuan National Park (LHZX-2022-03), the CAS “Light of West China” Program (2024), and the leading talents of the Kunlun talents in Qinghai Province (2024).
Author information
Authors and Affiliations
Contributions
Conceptualization: Faqi Zhang, Shilong Chen; Experimental design and methodology: Shuang Han, Yu Niu; Sample collection and laboratory work: Yu Niu, Yun Han; Data analysis: Shuang Han (genome assembly, gene annotation, SNP calling, population genetics analyses), Jingya Yu (functional annotation, enrichment analysis), Hao Xu (statistical analysis, data visualization); Visualization and figure preparation: Shuang Han, Hao Xu; Writing-original draft: Shuang Han; Writing-review & editing: All authors; Supervision and project administration: Faqi Zhang, Shilong Chen; Funding acquisition: Faqi Zhang, Shilong Chen. All authors have read and approved the final manuscript. The authors declare that they have no competing financial interests or personal relationships that could have influenced the work reported.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shuang, H., Niu, Y., Xu, H. et al. A High-quality chromosome-level genome assembly of Swertia przewalskii Pissjauk. Sci Data 12, 2026 (2025). https://doi.org/10.1038/s41597-025-06292-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06292-y
