
Abstract
The order Siluriformes, a hyperdiverse teleost clade with over 3,000 species, exemplifies adaptive radiation through significant phenotypic innovations such as aerial respiration. However, the limited availability of high-quality genomes, particularly in underrepresented families such as Sisoridae, has hindered phylogenomic and mechanistic studies of their ecological diversification. Here, we present a nearly complete telomere-to-telomere (T2T) genome assembly of Bagarius rutilus (Banded Goonch) generated by a hybrid sequencing approach integrating PacBio HiFi, Oxford Nanopore ultra-long and Hi-C technologies. The 618.3 Mb genome (contig N50 = 21.06 Mb, scaffold N50 = 25.46 Mb) was resolved into 26 chromosomes, achieving a genome coverage of 98.17%. The quality of the assembly was validated by a BUSCO score of 97.5% (Actinopterygii_odb10) and high conservation of synteny with other catfish species. Based on RNA sequencing, homology-based and de novo annotation, we identified 29,106 protein-coding genes. This chromosome-level genome represents one of the highest quality Siluriformes assemblies to date and provides a critical resource for reconstructing adaptive landscapes and exploring the genetic basis of phenotypic innovation in catfish.
Similar content being viewed by others


Background & Summary
The order Siluriformes (catfishes) is a quintessential model to understand the adaptive radiation in teleost fishes, comprising over 3,000 species (~25,000 protein coding genes) in more than 36 families that exhibit extraordinary phenotypic innovations1. These include the suprabranchial labyrinth organ in Clariidae for aerial respiration2, muscle-derived high-voltage bioelectric organs (>500 V) in Malapteruridae (electric catfish)3, keratinized feeding structures in Loricariidae (suckermouth armored catfish)4, and specialized hematophagous oral apparatus in parasitic lineages5. Such structural and functional innovations have enabled their colonization of diverse aquatic niches, ranging from freshwater to brackish ecosystems, and including benthic detritivory, filter feeding, blood parasitism, and even cave-dwelling adaptations3,4,5,6. Together, these features position Siluriformes as an unparalleled system for studying the mechanisms of adaptive radiation in teleosts.
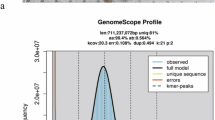
Despite significant advances in catfish biology7,8,9, critical gaps in genomic resources remain, particularly within some families such as the Sisoridae, hindering comprehensive phylogenomic frameworks and mechanistic insights into their adaptive evolution. In this study we focused on a representative Sisoridae species Bagarius rutilus (banded goonch; Fig. 1A), which has restricted distribution ranges in Laos, Vietnam and China. In addition, this species is a second-class protected species in China and has important protection value. We present a near complete telomere-to-telomere (T2T) genome assembly of banded goonch using a multiple sequencing strategy with HiFi, ONT ultra-long and Hi-C technologies. The assembled genome spans approximately 618 megabase pairs (Mb) across 26 chromosomes (contig N50 = 21.06 Mb, scaffold N50 = 25.46 Mb) (Fig. 1B,C). Seventeen chromosomes are completely free of gaps, six contain a single gap, and the remaining chromosomes contain more than one (Fig. 1D,E).

Chromosome-level genome assembly of the banded goonch (Bagarius rutilus). (A) The picture of the banded goonch. (B) Summary of genome characteristics including estimated genome size and heterozygosity. (C) Hi-C interaction heatmap showing chromosomal compartmentalization and spatial proximity. Chromosomes 3, 4, 25 and 26 are magnified to highlight intrachromosomal interaction patterns. (D) Distribution of contig counts per chromosome, with regions to the left of the red line representing gap-free assemblies. (E) Ideogram of the banded goonch karyotype. Blue triangles at chromosome ends indicate putative telomeric regions, and orange rectangles indicate residual assembly gaps.
High continuity and accuracy were confirmed by the completeness of conserved core genes (BUSCO score: 97.5%) and syntenic alignment with related species (Trichomycterus rosablanca). This assembly represents one of the highest quality Siluriformes genomes, providing a critical genomic resource for resolving long-standing phylogenetic uncertainties10 and elucidating the genetic basis of ecological specialization and protecting this species in the future.
Sample collection
A female Bagarius rutilus specimen was collected from Gasa Town, Xinping County, Yuxi City, Yunnan Province, China and ethically anesthetized with MS-222 (tricaine methanesulfonate) prior to dissection as previous study11. Skeletal muscle, heart, kidney and liver tissues were rapidly sampled, snap frozen in liquid nitrogen to preserve macromolecular integrity, and stored at −80 °C for subsequent genomic and transcriptomic analyses. Skeletal muscle was subjected to multi-platform sequencing, including short-read, high-fidelity long-read (PacBio Sequel II) and ultra-long nanopore technologies to ensure high quality genome assembly, while liver tissue was subjected to Hi-C sequencing for chromosome-scale scaffolding. In addition, RNA sequencing was performed on skeletal muscle, heart, kidney and liver tissues to construct a reference transcriptome and validate gene models.
Genomic HiFi sequencing
High molecular weight DNA was extracted from skeletal muscle using the MagAttract HMW DNA Kit (Qiagen) with optimized protocols: tissue homogenisation in ice-cold lysis buffer (2% β-mercaptoethanol) using a Potter-Elvehjem homogeniser (30 strokes at 200 rpm), extended Proteinase K digestion (18 h at 56 °C with 15 rpm rotation) and MinElute column purification with pre-warmed EB buffer (65 °C). For SMRTbell library construction, genomic DNA was mechanically sheared into 15–20 kb fragments using g-TUBE (Covaris; 3 × 6,000 × g pulses), followed by dual enzyme damage repair (NEBNext FFPE: PreCR = 1:3, 37 °C 60 min) and BluePippin size selection (0.75% agarose, 15–25 kbp narrow window). Ligation efficiency was validated by SMRTbell specific qPCR (Kapa Biosystems). PacBio Sequel II sequencing in CCS mode (30 hours run time) included polymerase load optimisation (Binding Calculator v6.0) and magnetic bead enrichment (Sequel II Binding Kit 3.0) targeting 80% ZMW occupancy, resulting in 137 Gb HiFi reads (average 17.3 kbp) for downstream analyses.
ONT Ultra-long sequencing
Ultra-long DNA was extracted using an adapted protocol from the Nanobind CBB Big DNA Kit (Circulomics, #102-301-900), where tissue was embedded in low melting point agarose (1% in PBS) and subjected to proteinase K digestion (2 mg/mL, 50 °C, 72 h). DNA was quantified using Qubit 4.0 (Invitrogen) and size profiled using the Femto Pulse System (Agilent). Library preparation was performed using the LSK-114 Ligation Sequencing Kit (Oxford Nanopore, #SQK-LSK114) with custom modifications, including DNA repair using Ultra II End-prep enzyme mix supplemented with 5% PEG 8000 (w/v), extended adapter ligation (30 min) with 1.5 × adapter concentration, and loading onto R10.4.1 flow cells pre-equilibrated with ELB supplemented with 0.05% Tween-20. Sequencing was performed on a PromethION P48 platform using MinKNOW v22.10.7 with adaptive sampling disabled, followed by basecalling using the Super Accurate (SUP) model in Dorado v0.3.0 and 49.57 Gb reads were generated.
Short and Hi-C reads sequencing
For short-read whole-genome sequencing, high-quality genomic DNA was sheared to an average fragment size of ~350 bp using a Covaris S220 system. A paired-end library was constructed following the standard protocol of the MGIEasy FS DNA Library Prep Kit (MGI Tech) and subsequently sequenced on a DNBSEQ-T7 platform in paired-end 150 bp (PE150) mode.
Hi-C library preparation and sequencing fresh skeletal muscle tissue was cross-linked in 1% formaldehyde for 10 min at room temperature, followed by quenching with 0.125 M glycine. Cell nuclei were isolated and lysed, and chromatin was digested with the methylation-sensitive restriction enzyme DpnII at 37 °C for 4 h to generate cohesive ends. Digested DNA fragments were biotinylated with biotin-14-dCTP and subjected to proximity ligation using T4 DNA ligase at 16 °C for 4 hours. Ligated DNA was purified, sheared to 300–500 bp fragments using a Covaris S220 and enriched for biotinylated junctions by streptavidin bead capture. Libraries were constructed using the MGIEasy FS DNA Library Prep Kit, with fragment size distribution validated using the Agilent 2100 Bioanalyzer, and sequenced on a DNBSEQ-T7 platform (MGI Tech) in paired-end 150 bp mode.
RNA sequencing
Total RNA was extracted from tissue samples using TRIzol reagent (Invitrogen, 15596026) according to the manufacturer’s protocol, followed by DNase I (NEB, #M0303) treatment to remove genomic DNA contamination. RNA quality and integrity were assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies) to ensure an RNA Integrity Number (RIN) ≥ 7.0 for library construction.
Poly(A) + mRNA was enriched using oligo (dT) magnetic beads and subsequently fragmented into ~250 bp fragments. First-strand cDNA was synthesized using random hexamer primers and reverse transcriptase, followed by second-strand cDNA synthesis. The resulting cDNA was purified, end repaired, A-tailed and ligated to MGI-specific adaptors. After ligation, the cDNA was amplified by PCR using a limited number of cycles to minimize bias. The final libraries were assessed for size distribution using the Agilent 2100 Bioanalyzer and quantified using Qubit 4.0 (Invitrogen).
Chromosome level genome assembly
Initial genome characterization was performed by K-mer analysis using Jellyfish (v2.3.1; k-mer size = 33)12 and GenomeScope (v2.0)13, which estimated a haploid genome size of 622 Mb with 0.3% heterozygosity (Fig. 1A–D). To ensure high quality input data for assembly, PacBio HiFi reads (≥Q20 accuracy, 15–20 kb) and Oxford Nanopore Technologies (ONT) ultra-long reads (N50 > 100 kb) underwent rigorous pre-processing: HiFi reads were filtered with HiFiAdapterFilt14 to remove adapter sequences, while ONT reads were quality trimmed with NanoFilt15 (minimum Q score = 7, minimum length = 50 kb).
A hybrid assembly strategy integrating HiFi, ONT ultra-long and Hi-C reads was used to achieve chromosome-scale continuity. Primary contigs were assembled from HiFi and ONT ultra-long reads using Hifiasm (v0.24.0-r702)16, using HiFi accuracy for base-level resolution and ONT reads to resolve long repetitive regions. Chromosome scaffolding was then performed using Hi-C data: reads were aligned to the draft assembly using BWA (v0.7.12)17, and chromatin interaction patterns were used to iteratively correct scaffold orientation and linkage using yahs (v1.2a.1)18. This step anchored 98% of the assembly into chromosome-scale pseudomolecules. Finally, we used quarTeT19 (v1.2.5) to fill gaps based on the HiFi and ONT reads, and the telomeres were also identified by quarTeT (Fig. 1D).
The completeness and accuracy of the assembly was validated using BUSCO (v5.5.0; actinopterygii_odb10)20, which identified 97.5% of conserved actinopterygii genes, confirming a comprehensive representation of gene space. Spatial consistency and chromosomal integrity were further verified by generating a Hi-C contact matrix with Juicer (v1.6)21 and visualising interaction patterns in Juicebox (v1.11.08)22 (Fig. 1B). Finally, the assembled genome size was approximately 618 Mb across 26 chromosomes. Of these, 17 were completely gap-free, six contained only a single gap, and three had more than one gaps (Fig. 1B,C and Table 1).
HiFi reads depth calculation
HiFi reads were aligned to the reference genome using Minimap2 (v2.25)23 with the -ax map-hifi option. The resulting BAM file was then used to calculate the sequencing depth at each base position using the bedtools (v2.25.0)24 genomecov utility. To quantify the distribution of reads along each chromosome, we computed the average read depth across consecutive, non-overlapping 100 kb windows using the bedtools make windows and map utilities. This process yielded the mean coverage for each 100 kb genomic bin, enabling the visualisation of read distribution patterns at the chromosomal level (Fig. 2).

Distribution of HiFi read sequencing depth across each chromosome. Each point represents the mean sequencing depth calculated in a 100 kb window.
Annotation of genome assembly
Repetitive elements in the banded goonch genome assembly were annotated through a combination of homology-based and de novo approaches. Homology detection was performed using RepeatProteinMask25 for protein-level alignment of transposable elements (TEs) and RepeatMasker26 for DNA-level identification. Tandem repeats were predicted with TRF27 using parameters optimized for sensitivity (trf 2 7 7 80 10 50 2000 -d -h). To capture species-specific repeats, a custom repeat library was constructed using RepeatModeler (v1.73)25, which generated consensus sequences through iterative classification. Non-overlapping annotations from homology-based, tandem repeat, and de novo predictions were merged into a unified dataset using bedtools (v2.25.0)24, resolving redundant overlaps. Collectively, repetitive elements comprised 26.70% of the genome assembly (Table 2).
Protein-coding genes were annotated using an integrative approach that combined ab initio prediction, homology-based evidence, and transcriptomic support, as in previous studies28,29,30. RNA-seq data from skeletal muscle, heart, kidney and liver were assembled using rnaSPAdes (v3.13.1)31, and putative coding sequences were identified from the assembled transcripts using TransDecoder (v5.5.0)32. For homology-based prediction, protein sequences from seven close species (Ictalurus punctatus33 [GCA_001660625.3], Ictalurus furcatus34 [GCA_023375685.2], Hemibagrus wyckioides35 [GCA_019097595.1], Silurus meridionalis36 [GCA_014805685.1], Clarias dussumieri37 [GCA_041682065.1]. Finally, EVidenceModeler (v1.1.1)38 integrated predictions from all three methods, prioritizing transcriptome-derived evidence (weight = 10), followed by homology (weight = 5) and ab initio predictions (weight = 1), yielding a consensus set of 29,106 protein-coding genes with high confidence.
Syntenic alignment with Trichomycterus rosablanca
The genome of Trichomycterus rosaceus39 (assembly accession: GCA_030014385.1) was retrieved from the National Center for Biotechnology Information (NCBI) database for the comparative syntenic analysis. The protein-coding genes within this genome were then annotated using the aforementioned genome annotation pipeline. This annotated genome was then used to perform a whole-genome synteny comparison with our target species using JCVI40. To refine the results, the identified syntenic blocks were filtered based on two criteria: (1) blocks containing fewer than five genes were discarded and (2) only syntenic gene pairs with sequence identities greater than 75% were retained for subsequent analysis (Fig. 3A,B).

Comparative synteny and genomic architecture of the banded goonch (Bagarius rutilus). (A) Circos plot illustrates collinear relationships between the banded goonch (blue tracks) and the Trichomycterus rosablanca (green tracks). Connecting ribbons represent conserved homologous regions. (B) Circular visualization of genomic features across the banded goonch genome, analyzed in 500-kb non-overlapping windows. The color scale of the heatmap represents the Z-score normalized values for each data track. a: chromosomes, b: gene frequency across the genome, c: density of DNA transposable elements, d: density of long interspersed nuclear elements (LINEs), e: density of long terminal repeats (LTRs), f: density of short interspersed nuclear elements (SINEs), g: density of other transposable elements, h: density of unknown genomic elements, i: the collinearity relationships within genome.
Data Records
HiFi, ONT ultra-long, Hi-C and RNA-seq raw reads were deposited in the China National GeneBank Database (CNGBdb) under the accession numbers CNP000705841,42,43,44,45,46,47,48,49. The genome assembly and annotation were also can be found in figshare database (https://figshare.com/articles/dataset/Genome_genome_annotation_information_and_protein_sequence_of_Bagarius_rutilus/28660733)50,51 and GenBank (JBRFMU000000000.1)52.
Technical Validation
The integrity of the genomic DNA was verified by agarose gel electrophoresis and its concentration quantified using a Qubit 4.0 Fluorometer with the 1 × dsDNA High Sensitivity (HS) Assay Kit (Thermo Fisher Scientific). The final genome assembly demonstrated a high degree of completeness, as evidenced by a BUSCO score of 97.5%, and exhibited strong syntenic collinearity with the genome of the related catfish species Trichomycterus rosaceus. To further assess the accuracy and contiguity of the assembly, the raw sequencing reads were mapped back, yielding high alignment rates and coverage depths: 99.71% for HiFi reads (at 223 × mean coverage), 99.85% for ONT ultra-long reads (at 41 × mean coverage), and 100% for Hi-C reads, confirming the high quality of the final genome assembly.
Data availability
All raw sequencing data including HiFi, ONT ultra-long, Hi-C and RNA-seq raw reads and genome assembly were deposited in the China National GeneBank Database (CNGBdb) under the accession numbers CNP000705841,42,43,44,45,46,47,48,49, which includes Illumina short reads (CNX1195290)41, Hi-C data (CNX1195291)42, ONT ultra-long reads (CNX1195292)43, PacBio HiFi reads (CNX1195293)44, RNA-seq data (CNX1195294–CNX1195297)45,46,47,48 and genome assembly (CNA0423406)49. The genome assembly and annotation information were also can be found in figshare database50,51 and GenBank (JBRFMU000000000.1)52.
Code availability
No specific script was used in this work. The corresponding bioinformatics software and the specific versions of software have been described in Methods.
References
Segaran, T. et al. Catfishes: A global review of the literature. Heliyon 9, e20081 (2023).
Mbanga, B., van Dyk, C. & Maina, J. N. Morphometric and morphological study of the respiratory organs of the bimodally-breathing African sharptooth catfish (Clarias gariepinus). Burchell (1822). Zoology 130, 6–18 (2018).
Welzel, G. & Schuster, S. Efficient high-voltage protection in the electric catfish. J Exp. Biol. 224, jeb239855 (2021).
Krings, W., Konn-Vetterlein, D., Hausdorf, B. & Gorb, S. N. Holding in the stream: convergent evolution of suckermouth structures in Loricariidae (Siluriformes). Front. Zool. 20, 37 (2023).
Henschel, E., Baskin, J. N., Collins, R. & Lujan, N. K. A revised diagnosis of the blood-feeding candiru genus Paravandellia (Siluriformes: Trichomycteridae: Vandelliinae) with descriptions of three new species. American Museum Novitates 2024, 1–36 (2024).
Armbruster, J. W. Global catfish biodiversity. in American Fisheries Society Symposium (2011).
Gutiérrez-Barragán, A., Varela-Romero, A., García-De León, F. J. & Grijalva-Chon, J. M. A review of catfish (Siluriformes) hybridization. Rev. Fish. Biol. Fisheries 35, 145–174 (2025).
Yang, L. et al. Enlarged fins of Tibetan catfish provide new evidence of adaptation to high plateau. Sci. China Life Sci. 66, 1554–1568 (2023).
Zhou, Q. et al. Decoding the fish genome opens a new era in important trait research and molecular breeding in China. Sci. China Life Sci. 67, 2064–2083 (2024).
Schedel, F. D. et al. New phylogenetic insights into the African catfish families Mochokidae and Austroglanididae. J Fish Biol. 100, 1171–1186 (2022).
Wu, B. et al. Single-cell analysis of the amphioxus hepatic caecum and vertebrate liver reveals genetic mechanisms of vertebrate liver evolution. Nat. Ecol. Evol. 8, 1972–1990 (2024).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Sim, S. B., Corpuz, R. L., Simmonds, T. J. & Geib, S. M. HiFiAdapterFilt, a memory efficient read processing pipeline, prevents occurrence of adapter sequence in PacBio HiFi reads and their negative impacts on genome assembly. BMC Genomics 23, 157 (2022).
De Coster, W., D’hert, S., Schultz, D. T., Cruts, M. & Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669 (2018).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Li, H. & Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39, btac808 (2023).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Hortic. Res. 10, uhad127 (2023).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Saha, S., Bridges, S., Magbanua, Z. V. & Peterson, D. G. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 36, 2284–94 (2008).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25, 4–10 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Wu, B. et al. The genomes of two billfishes provide insights into the evolution of endothermy in teleosts. Mol. Biol. Evol. 38, 2413–2427 (2021).
Wu, B. et al. Distinct and shared endothermic strategies in the heat producing tissues of tuna and other teleosts. Sci. China Life Sci. 66, 629–2645 (2023).
Wu, B. et al. Resequencing of reindeer genomes provides clues to their docile habits. Evol. Lett. 8, 494–504 (2024).
Bushmanova, E., Antipov, D., Lapidus, A. & Prjibelski, A. D. rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 8, giz100 (2019).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–512 (2013).
Chen, X. et al. High-quality genome assembly of channel catfish, Ictalurus punctatus. GigaScience 5, 39 (2016).
Wang, H., Su, B., Butts, I. A. E., Dunham, R. A. & Wang, X. Chromosome-level assembly and annotation of the blue catfish Ictalurus furcatus, an aquaculture species for hybrid catfish reproduction, epigenetics, and heterosis studies. GigaScience 11, giac070 (2022).
Shao, F. et al. Chromosome-Level Genome Assembly of the Asian Red-Tail Catfish (Hemibagrus wyckioides). Front. Genet. 12, 747684 (2021).
Zheng, S. et al. Chromosome-level assembly f southern catfish (Silurus meridionalis) provides insights into visual adaptation to nocturnal and benthic lifestyles. Mol. Ecol. Resour. 21, 1575–1592 (2021).
Mohindra, V., Chowdhury, L. M., Charan, R., Basheer, V. S. & Jena, J. Genome sequencing and assembly of near threatened Clarias dussumieri (Valenciennes,1840), an endemic catfish of peninsular India. Sci. Data. 11, 1406 (2024).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using Evidence modeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Cadena, C. D. et al. A reference genome for the Andean cavefish Trichomycterus rosablanca (Siluriformes, Trichomycteridae): Building genomic resources to study evolution in cave environments. J Hered. 115, 311–316 (2024).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488 (2008).
short reads genome sequence of Bagarius rutilus, Genome Sequence Archive, https://db.cngb.org/search/?q=CNX1195290 (2025).
Hi-C sequence of Bagarius rutilus, Genome Sequence Archive, https://db.cngb.org/search/?q=CNX1195291 (2025).
ONT ultra-long sequence of Bagarius rutilus, Genome Sequence Archive, https://db.cngb.org/search/?q=CNX1195292 (2025).
HIFI sequence of Bagarius rutilus, Genome Sequence Archive, https://db.cngb.org/search/?q=CNX1195293 (2025).
RNA-Seq of Bagarius rutilus:brain trans_brain, Genome Sequence Archive, https://db.cngb.org/search/?q=CNX1195294 (2025).
RNA-Seq of Bagarius rutilus:heart trans_heart, Genome Sequence Archive, https://db.cngb.org/search/?q=CNX1195295 (2025).
RNA-Seq of Bagarius rutilus:kidney trans_kidney, Genome Sequence Archive, https://db.cngb.org/search/?q=CNX1195296 (2025).
RNA-Seq of Bagarius rutilus:liver trans_liver, Genome Sequence Archive, https://db.cngb.org/search/?q=CNX1195297 (2025).
Bagarius_rutilus_fas, Genome Sequence Archive, https://db.cngb.org/search/?q=CNA0423406 (2025).
Repeat sequence information of the Bagarius rutilus genome, figshare, https://doi.org/10.6084/m9.figshare.28681337.v1.
Genome, genome annotation information, and protein sequence of Bagarius rutilus, figshare, https://doi.org/10.6084/m9.figshare.28660733.v1.
NCBI Assembly. https://identifiers.org/ncbi/insdc.gca:GCA_053477655.1 (2025).
Acknowledgements
This work was supported by the Project of Finance Special Fund of Chinese Ministry of Agriculture and Rural Affairs of the People’s Republic of China-Fisheries Resources and Environment Survey in the key water areas of Southwest China (2023–2025), he National Natural Science Foundation of China (Grant No.32503195), Scientific Innovation Fund of Pearl River Fisheries Research Institute, Chinese Academy of Fishery Science (2023CXYC6, 2024CXYC5), Innovation Team of Survey and Assessment of the Pearl River Fishery Resources (2023TD-10), Finance Special Fund of Chinese Ministry of Agriculture and Rural Affairs of the People’s Republic of China-Fishery Resources and Habitat Survey in the Pearl River Basin (2024–2026).
Author information
Authors and Affiliations
Contributions
Y.L., F.Z., J.L., M.D., J.L. and W.C. conceived and designed the project. C.L., H.Y. and Y.L. collected the samples. Y.L. and F.Z. performed the DNA and RNA extraction, library preparation, and genome sequencing. W.C. performed the bioinformatics analysis and visualized the results. Y.L., F.Z., J.L., M.D., J.L. and W.C. wrote the manuscript. Y.L. and W.C. revised and edited the manuscript. All authors have read and approved the final version of manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Y., Zhu, F., Lei, C. et al. Near complete T2T genome assembly of the banded goonch (Bagarius rutilus). Sci Data 13, 91 (2026). https://doi.org/10.1038/s41597-025-06403-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06403-9
